 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEstimating a common covariance matrix for network meta-analysis of gene expression datasets in diffuse large B-cell lymphoma
Aug 21, 2017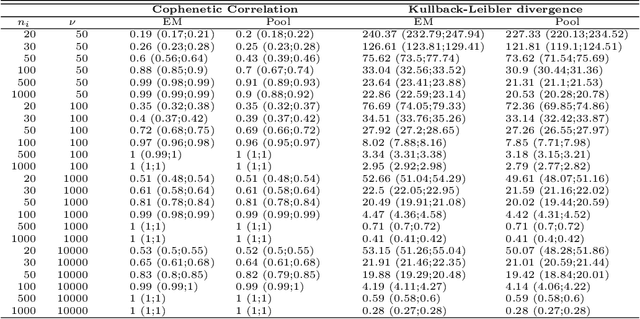

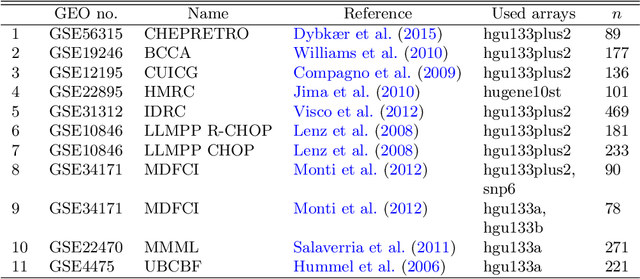
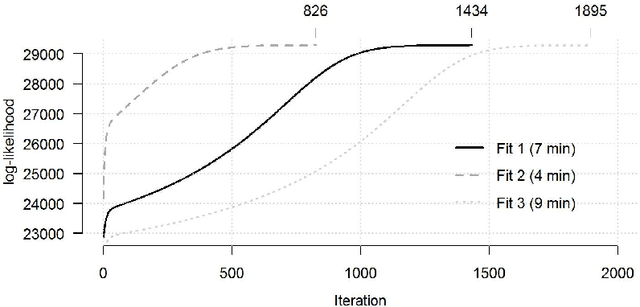
The estimation of covariance matrices of gene expressions has many applications in cancer systems biology. Many gene expression studies, however, are hampered by low sample size and it has therefore become popular to increase sample size by collecting gene expression data across studies. Motivated by the traditional meta-analysis using random effects models, we present a hierarchical random covariance model and use it for the meta-analysis of gene correlation networks across 11 large-scale gene expression studies of diffuse large B-cell lymphoma (DLBCL). We suggest to use a maximum likelihood estimator for the underlying common covariance matrix and introduce an EM algorithm for estimation. By simulation experiments comparing the estimated covariance matrices by cophenetic correlation and Kullback-Leibler divergence the suggested estimator showed to perform better or not worse than a simple pooled estimator. In a posthoc analysis of the estimated common covariance matrix for the DLBCL data we were able to identify novel biologically meaningful gene correlation networks with eigengenes of prognostic value. In conclusion, the method seems to provide a generally applicable framework for meta-analysis, when multiple features are measured and believed to share a common covariance matrix obscured by study dependent noise.