 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVAE-MS: An Asymmetric Variational Autoencoder for Mutational Signature Extraction
Feb 24, 2026Mutational signature analysis has emerged as a powerful method for uncovering the underlying biological processes driving cancer development. However, the signature extraction process, typically performed using non-negative matrix factorization (NMF), often lacks reliability and clinical applicability. To address these limitations, several solutions have been introduced, including the use of neural networks to achieve more accurate estimates and probabilistic methods to better capture natural variation in the data. In this work, we introduce a Variational Autoencoder for Mutational Signatures (VAE-MS), a novel model that leverages both an asymmetric architecture and probabilistic methods for the extraction of mutational signatures. VAE-MS is compared to with three state-of-the-art models for mutational signature extraction: SigProfilerExtractor, the NMF-based gold standard; MUSE-XAE, an autoencoder that employs an asymmetric design without probabilistic components; and SigneR, a Bayesian NMF model, to illustrate the strength in combining a nonlinear extraction with a probabilistic model. In the ability to reconstruct input data and generalize to unseen data, models with probabilistic components (VAE-MS, SigneR) dramatically outperformed models without (SigProfilerExtractor, MUSE-XAE). The NMF-baed models (SigneR, SigProfilerExtractor) had the most accurate reconstructions in simulated data, while VAE-MS reconstructed more accurately on real cancer data. Upon evaluating the ability to extract signatures consistently, no model exhibited a clear advantage over the others. Software for VAE-MS is available at https://github.com/CLINDA-AAU/VAE-MS.
On the Relation Between Autoencoders and Non-negative Matrix Factorization, and Their Application for Mutational Signature Extraction
May 13, 2024



The aim of this study is to provide a foundation to understand the relationship between non-negative matrix factorization (NMF) and non-negative autoencoders enabling proper interpretation and understanding of autoencoder-based alternatives to NMF. Since its introduction, NMF has been a popular tool for extracting interpretable, low-dimensional representations of high-dimensional data. However, recently, several studies have proposed to replace NMF with autoencoders. This increasing popularity of autoencoders warrants an investigation on whether this replacement is in general valid and reasonable. Moreover, the exact relationship between non-negative autoencoders and NMF has not been thoroughly explored. Thus, a main aim of this study is to investigate in detail the relationship between non-negative autoencoders and NMF. We find that the connection between the two models can be established through convex NMF, which is a restricted case of NMF. In particular, convex NMF is a special case of an autoencoder. The performance of NMF and autoencoders is compared within the context of extraction of mutational signatures from cancer genomics data. We find that the reconstructions based on NMF are more accurate compared to autoencoders, while the signatures extracted using both methods show comparable consistencies and values when externally validated. These findings suggest that the non-negative autoencoders investigated in this article do not provide an improvement of NMF in the field of mutational signature extraction.
Regression on imperfect class labels derived by unsupervised clustering
Aug 16, 2019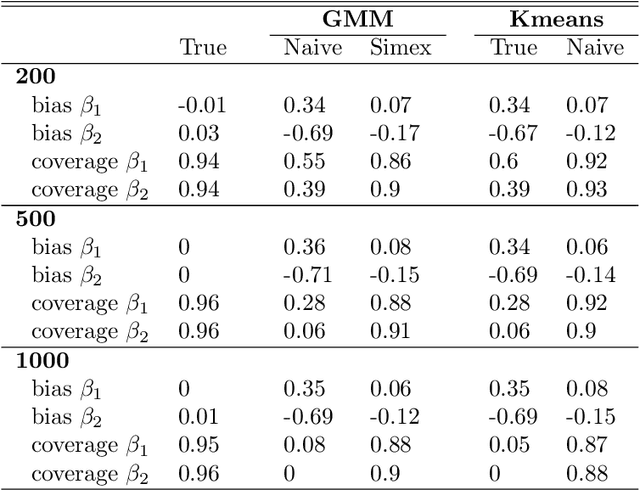
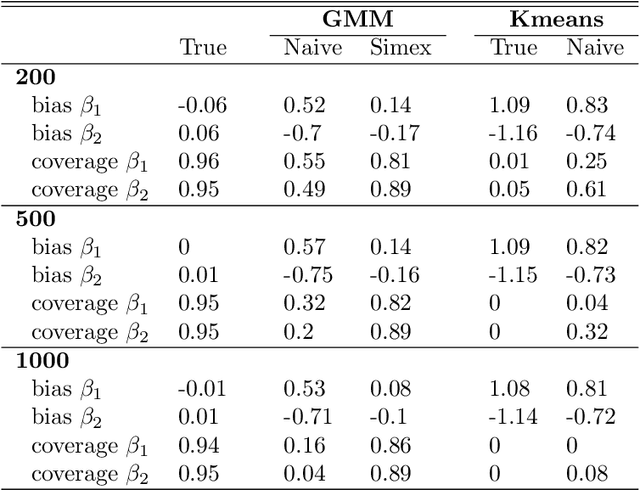
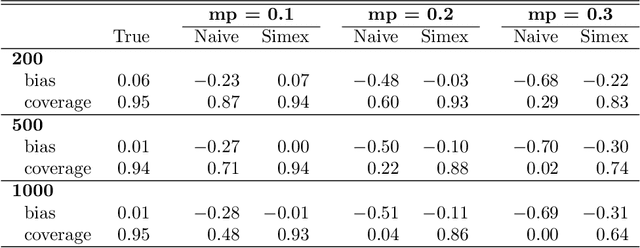
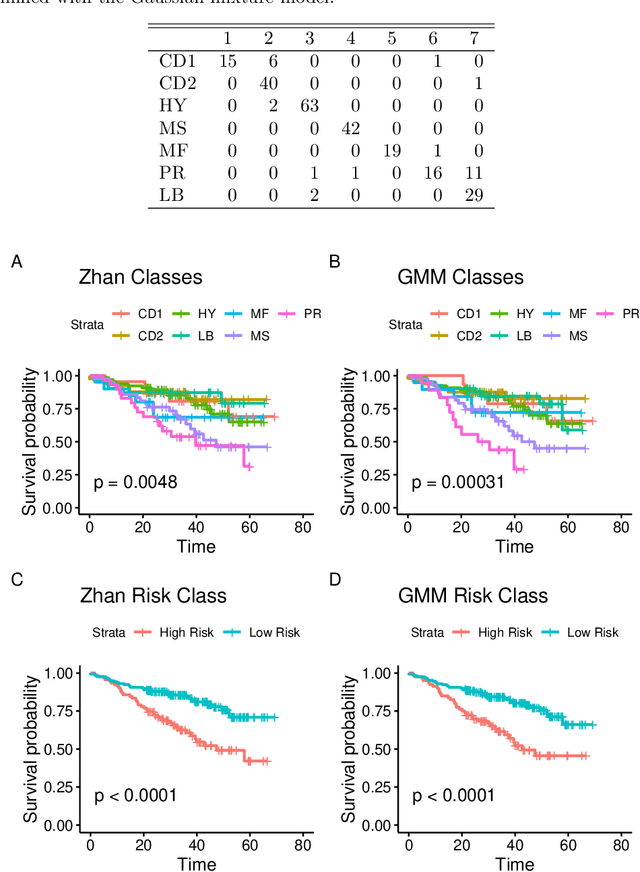
Outcome regressed on class labels identified by unsupervised clustering is custom in many applications. However, it is common to ignore the misclassification of class labels caused by the learning algorithm, which potentially leads to serious bias of the estimated effect parameters. Due to its generality we suggest to redress the situation by use of the simulation and extrapolation method. Performance is illustrated by simulated data from Gaussian mixture models. Finally, we apply our method to a study which regressed overall survival on class labels derived from unsupervised clustering of gene expression data from bone marrow samples of multiple myeloma patients.
Estimating a common covariance matrix for network meta-analysis of gene expression datasets in diffuse large B-cell lymphoma
Aug 21, 2017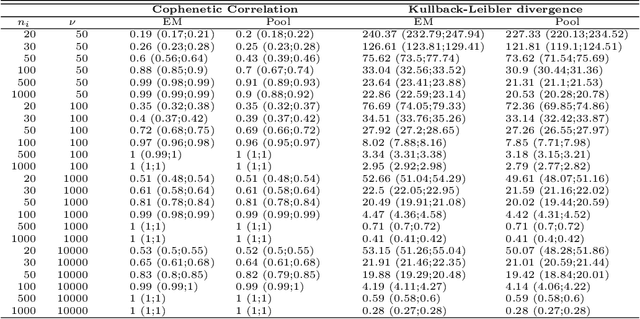

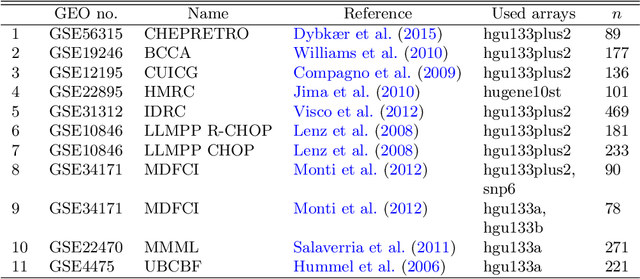
The estimation of covariance matrices of gene expressions has many applications in cancer systems biology. Many gene expression studies, however, are hampered by low sample size and it has therefore become popular to increase sample size by collecting gene expression data across studies. Motivated by the traditional meta-analysis using random effects models, we present a hierarchical random covariance model and use it for the meta-analysis of gene correlation networks across 11 large-scale gene expression studies of diffuse large B-cell lymphoma (DLBCL). We suggest to use a maximum likelihood estimator for the underlying common covariance matrix and introduce an EM algorithm for estimation. By simulation experiments comparing the estimated covariance matrices by cophenetic correlation and Kullback-Leibler divergence the suggested estimator showed to perform better or not worse than a simple pooled estimator. In a posthoc analysis of the estimated common covariance matrix for the DLBCL data we were able to identify novel biologically meaningful gene correlation networks with eigengenes of prognostic value. In conclusion, the method seems to provide a generally applicable framework for meta-analysis, when multiple features are measured and believed to share a common covariance matrix obscured by study dependent noise.