 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExploration of Dark Chemical Genomics Space via Portal Learning: Applied to Targeting the Undruggable Genome and COVID-19 Anti-Infective Polypharmacology
Nov 23, 2021
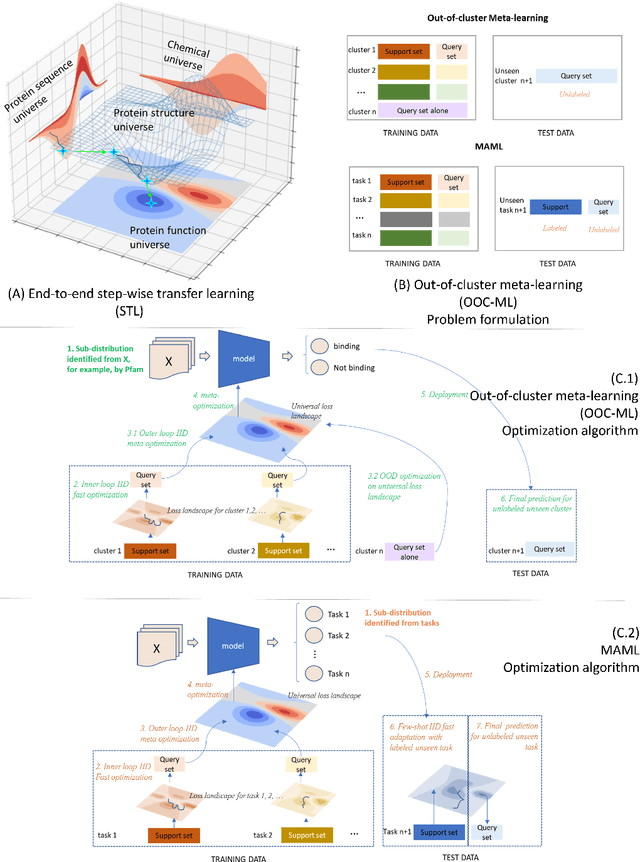
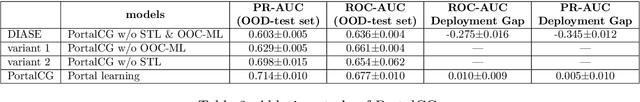
Advances in biomedicine are largely fueled by exploring uncharted territories of human biology. Machine learning can both enable and accelerate discovery, but faces a fundamental hurdle when applied to unseen data with distributions that differ from previously observed ones -- a common dilemma in scientific inquiry. We have developed a new deep learning framework, called {\textit{Portal Learning}}, to explore dark chemical and biological space. Three key, novel components of our approach include: (i) end-to-end, step-wise transfer learning, in recognition of biology's sequence-structure-function paradigm, (ii) out-of-cluster meta-learning, and (iii) stress model selection. Portal Learning provides a practical solution to the out-of-distribution (OOD) problem in statistical machine learning. Here, we have implemented Portal Learning to predict chemical-protein interactions on a genome-wide scale. Systematic studies demonstrate that Portal Learning can effectively assign ligands to unexplored gene families (unknown functions), versus existing state-of-the-art methods, thereby allowing us to target previously "undruggable" proteins and design novel polypharmacological agents for disrupting interactions between SARS-CoV-2 and human proteins. Portal Learning is general-purpose and can be further applied to other areas of scientific inquiry.
Machine Learning for Classification of Protein Helix Capping Motifs
May 01, 2019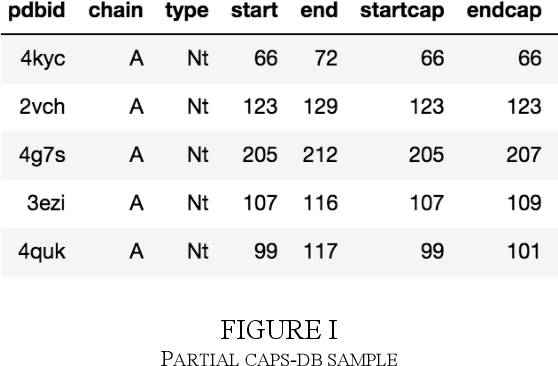
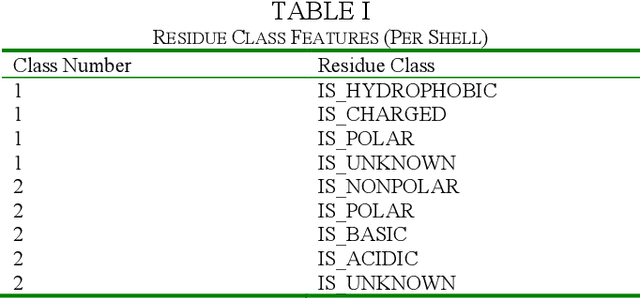
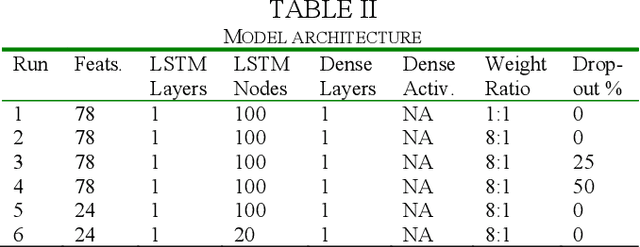
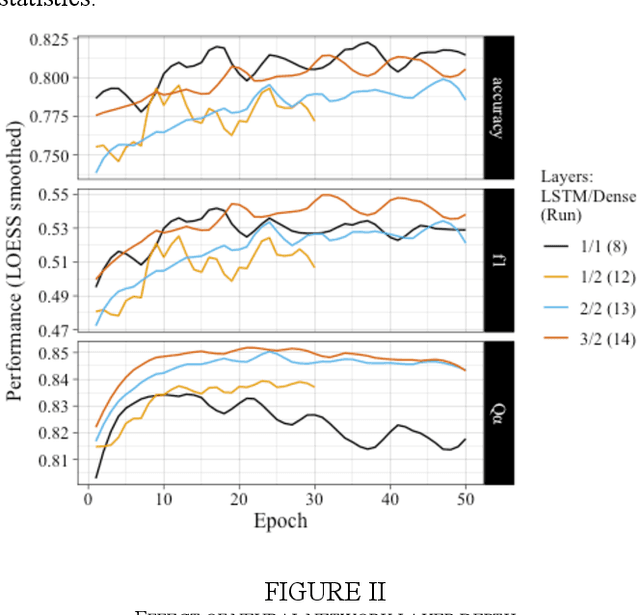
The biological function of a protein stems from its 3-dimensional structure, which is thermodynamically determined by the energetics of interatomic forces between its amino acid building blocks (the order of amino acids, known as the sequence, defines a protein). Given the costs (time, money, human resources) of determining protein structures via experimental means such as X-ray crystallography, can we better describe and compare protein 3D structures in a robust and efficient manner, so as to gain meaningful biological insights? We begin by considering a relatively simple problem, limiting ourselves to just protein secondary structural elements. Historically, many computational methods have been devised to classify amino acid residues in a protein chain into one of several discrete secondary structures, of which the most well-characterized are the geometrically regular $\alpha$-helix and $\beta$-sheet; irregular structural patterns, such as 'turns' and 'loops', are less understood. Here, we present a study of Deep Learning techniques to classify the loop-like end cap structures which delimit $\alpha$-helices. Previous work used highly empirical and heuristic methods to manually classify helix capping motifs. Instead, we use structural data directly--including (i) backbone torsion angles computed from 3D structures, (ii) macromolecular feature sets (e.g., physicochemical properties), and (iii) helix cap classification data (from CAPS-DB)--as the ground truth to train a bidirectional long short-term memory (BiLSTM) model to classify helix cap residues. We tried different network architectures and scanned hyperparameters in order to train and assess several models; we also trained a Support Vector Classifier (SVC) to use as a baseline. Ultimately, we achieved 85% class-balanced accuracy with a deep BiLSTM model.