 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMachine Learning for Classification of Protein Helix Capping Motifs
May 01, 2019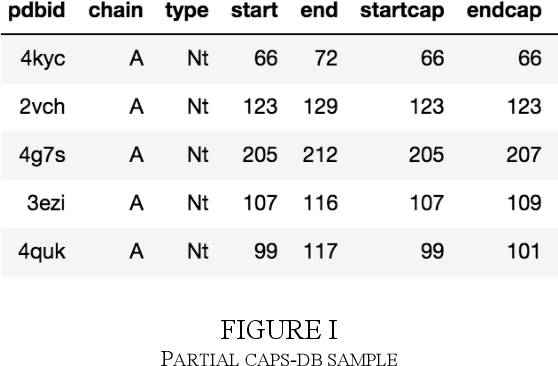
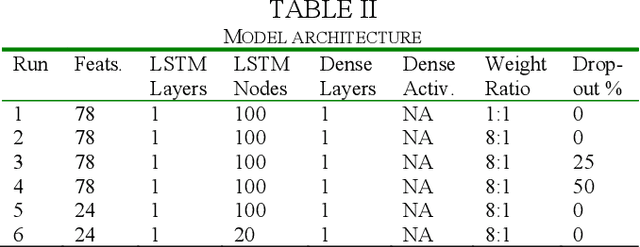
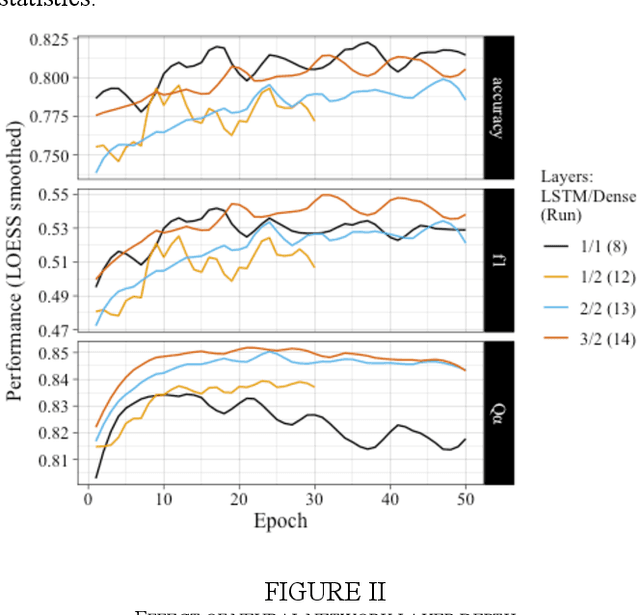
The biological function of a protein stems from its 3-dimensional structure, which is thermodynamically determined by the energetics of interatomic forces between its amino acid building blocks (the order of amino acids, known as the sequence, defines a protein). Given the costs (time, money, human resources) of determining protein structures via experimental means such as X-ray crystallography, can we better describe and compare protein 3D structures in a robust and efficient manner, so as to gain meaningful biological insights? We begin by considering a relatively simple problem, limiting ourselves to just protein secondary structural elements. Historically, many computational methods have been devised to classify amino acid residues in a protein chain into one of several discrete secondary structures, of which the most well-characterized are the geometrically regular $\alpha$-helix and $\beta$-sheet; irregular structural patterns, such as 'turns' and 'loops', are less understood. Here, we present a study of Deep Learning techniques to classify the loop-like end cap structures which delimit $\alpha$-helices. Previous work used highly empirical and heuristic methods to manually classify helix capping motifs. Instead, we use structural data directly--including (i) backbone torsion angles computed from 3D structures, (ii) macromolecular feature sets (e.g., physicochemical properties), and (iii) helix cap classification data (from CAPS-DB)--as the ground truth to train a bidirectional long short-term memory (BiLSTM) model to classify helix cap residues. We tried different network architectures and scanned hyperparameters in order to train and assess several models; we also trained a Support Vector Classifier (SVC) to use as a baseline. Ultimately, we achieved 85% class-balanced accuracy with a deep BiLSTM model.