 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMask-guided cross-image attention for zero-shot in-silico histopathologic image generation with a diffusion model
Jul 16, 2024



Creating in-silico data with generative AI promises a cost-effective alternative to staining, imaging, and annotating whole slide images in computational pathology. Diffusion models are the state-of-the-art solution for generating in-silico images, offering unparalleled fidelity and realism. Using appearance transfer diffusion models allows for zero-shot image generation, facilitating fast application and making model training unnecessary. However current appearance transfer diffusion models are designed for natural images, where the main task is to transfer the foreground object from an origin to a target domain, while the background is of insignificant importance. In computational pathology, specifically in oncology, it is however not straightforward to define which objects in an image should be classified as foreground and background, as all objects in an image may be of critical importance for the detailed understanding the tumor micro-environment. We contribute to the applicability of appearance transfer diffusion models to immunohistochemistry-stained images by modifying the appearance transfer guidance to alternate between class-specific AdaIN feature statistics matchings using existing segmentation masks. The performance of the proposed method is demonstrated on the downstream task of supervised epithelium segmentation, showing that the number of manual annotations required for model training can be reduced by 75%, outperforming the baseline approach. Additionally, we consulted with a certified pathologist to investigate future improvements. We anticipate this work to inspire the application of zero-shot diffusion models in computational pathology, providing an efficient method to generate in-silico images with unmatched fidelity and realism, which prove meaningful for downstream tasks, such as training existing deep learning models or finetuning foundation models.
Auxiliary CycleGAN-guidance for Task-Aware Domain Translation from Duplex to Monoplex IHC Images
Mar 12, 2024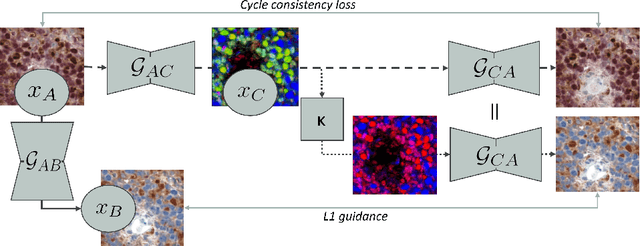

Generative models enable the translation from a source image domain where readily trained models are available to a target domain unseen during training. While Cycle Generative Adversarial Networks (GANs) are well established, the associated cycle consistency constrain relies on that an invertible mapping exists between the two domains. This is, however, not the case for the translation between images stained with chromogenic monoplex and duplex immunohistochemistry (IHC) assays. Focusing on the translation from the latter to the first, we propose - through the introduction of a novel training design, an alternative constrain leveraging a set of immunofluorescence (IF) images as an auxiliary unpaired image domain. Quantitative and qualitative results on a downstream segmentation task show the benefit of the proposed method in comparison to baseline approaches.
ReStainGAN: Leveraging IHC to IF Stain Domain Translation for in-silico Data Generation
Mar 11, 2024

The creation of in-silico datasets can expand the utility of existing annotations to new domains with different staining patterns in computational pathology. As such, it has the potential to significantly lower the cost associated with building large and pixel precise datasets needed to train supervised deep learning models. We propose a novel approach for the generation of in-silico immunohistochemistry (IHC) images by disentangling morphology specific IHC stains into separate image channels in immunofluorescence (IF) images. The proposed approach qualitatively and quantitatively outperforms baseline methods as proven by training nucleus segmentation models on the created in-silico datasets.
Stain Isolation-based Guidance for Improved Stain Translation
Jun 28, 2022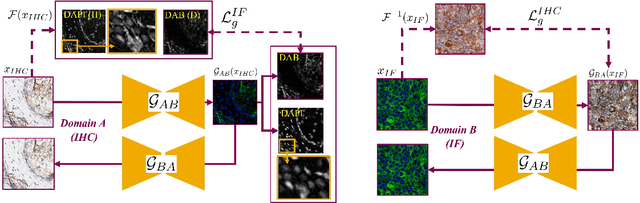
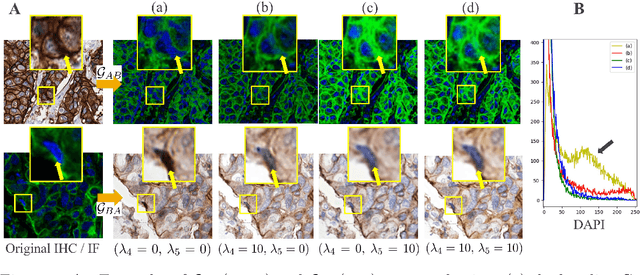
Unsupervised and unpaired domain translation using generative adversarial neural networks, and more precisely CycleGAN, is state of the art for the stain translation of histopathology images. It often, however, suffers from the presence of cycle-consistent but non structure-preserving errors. We propose an alternative approach to the set of methods which, relying on segmentation consistency, enable the preservation of pathology structures. Focusing on immunohistochemistry (IHC) and multiplexed immunofluorescence (mIF), we introduce a simple yet effective guidance scheme as a loss function that leverages the consistency of stain translation with stain isolation. Qualitative and quantitative experiments show the ability of the proposed approach to improve translation between the two domains.
Domain Adaptation-based Augmentation for Weakly Supervised Nuclei Detection
Jul 10, 2019
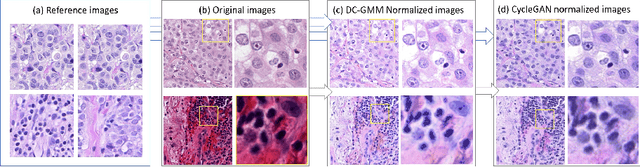
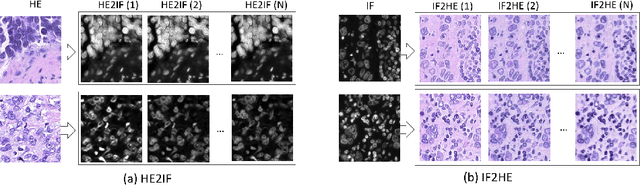
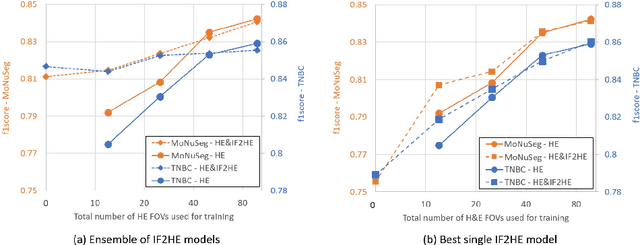
The detection of nuclei is one of the most fundamental components of computational pathology. Current state-of-the-art methods are based on deep learning, with the prerequisite that extensive labeled datasets are available. The increasing number of patient cohorts to be analyzed, the diversity of tissue stains and indications, as well as the cost of dataset labeling motivates the development of novel methods to reduce labeling effort across domains. We introduce in this work a weakly supervised 'inter-domain' approach that (i) performs stain normalization and unpaired image-to-image translation to transform labeled images on a source domain to synthetic labeled images on an unlabeled target domain and (ii) uses the resulting synthetic labeled images to train a detection network on the target domain. Extensive experiments show the superiority of the proposed approach against the state-of-the-art 'intra-domain' detection based on fully-supervised learning.
DASGAN -- Joint Domain Adaptation and Segmentation for the Analysis of Epithelial Regions in Histopathology PD-L1 Images
Jun 26, 2019



The analysis of the tumor environment on digital histopathology slides is becoming key for the understanding of the immune response against cancer, supporting the development of novel immuno-therapies. We introduce here a novel deep learning solution to the related problem of tumor epithelium segmentation. While most existing deep learning segmentation approaches are trained on time-consuming and costly manual annotation on single stain domain (PD-L1), we leverage here semi-automatically labeled images from a second stain domain (Cytokeratin-CK). We introduce an end-to-end trainable network that jointly segment tumor epithelium on PD-L1 while leveraging unpaired image-to-image translation between CK and PD-L1, therefore completely bypassing the need for serial sections or re-staining of slides. Extending the method to differentiate between PD-L1 positive and negative tumor epithelium regions enables the automated estimation of the PD-L1 Tumor Cell (TC) score. Quantitative experimental results demonstrate the accuracy of our approach against state-of-the-art segmentation methods.
Deep Semi Supervised Generative Learning for Automated PD-L1 Tumor Cell Scoring on NSCLC Tissue Needle Biopsies
Jun 28, 2018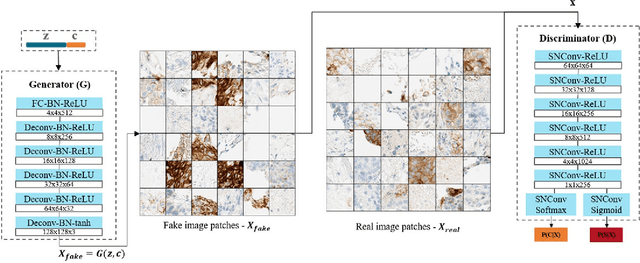
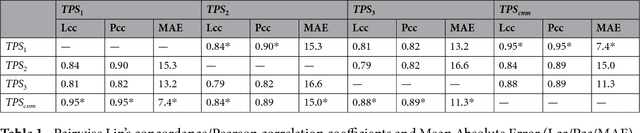

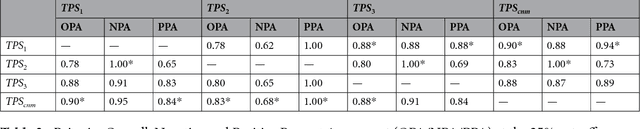
The level of PD-L1 expression in immunohistochemistry (IHC) assays is a key biomarker for the identification of Non-Small-Cell-Lung-Cancer (NSCLC) patients that may respond to anti PD-1/PD-L1 treatments. The quantification of PD-L1 expression currently includes the visual estimation of a Tumor Cell (TC) score by a pathologist and consists of evaluating the ratio of PD-L1 positive and PD-L1 negative tumor cells. Known challenges like differences in positivity estimation around clinically relevant cut-offs and sub-optimal quality of samples makes visual scoring tedious and subjective, yielding a scoring variability between pathologists. In this work, we propose a novel deep learning solution that enables the first automated and objective scoring of PD-L1 expression in late stage NSCLC needle biopsies. To account for the low amount of tissue available in biopsy images and to restrict the amount of manual annotations necessary for training, we explore the use of semi-supervised approaches against standard fully supervised methods. We consolidate the manual annotations used for training as well the visual TC scores used for quantitative evaluation with multiple pathologists. Concordance measures computed on a set of slides unseen during training provide evidence that our automatic scoring method matches visual scoring on the considered dataset while ensuring repeatability and objectivity.