 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeModeling nanoconfinement effects using active learning
May 07, 2020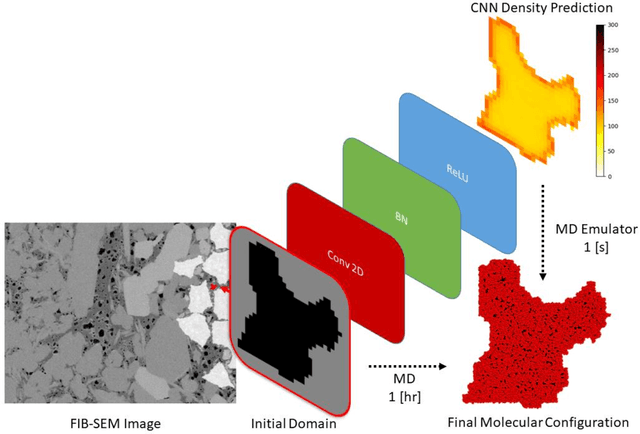

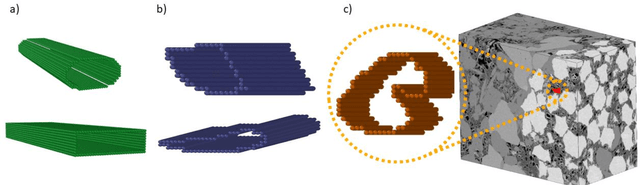

Predicting the spatial configuration of gas molecules in nanopores of shale formations is crucial for fluid flow forecasting and hydrocarbon reserves estimation. The key challenge in these tight formations is that the majority of the pore sizes are less than 50 nm. At this scale, the fluid properties are affected by nanoconfinement effects due to the increased fluid-solid interactions. For instance, gas adsorption to the pore walls could account for up to 85% of the total hydrocarbon volume in a tight reservoir. Although there are analytical solutions that describe this phenomenon for simple geometries, they are not suitable for describing realistic pores, where surface roughness and geometric anisotropy play important roles. To describe these, molecular dynamics (MD) simulations are used since they consider fluid-solid and fluid-fluid interactions at the molecular level. However, MD simulations are computationally expensive, and are not able to simulate scales larger than a few connected nanopores. We present a method for building and training physics-based deep learning surrogate models to carry out fast and accurate predictions of molecular configurations of gas inside nanopores. Since training deep learning models requires extensive databases that are computationally expensive to create, we employ active learning (AL). AL reduces the overhead of creating comprehensive sets of high-fidelity data by determining where the model uncertainty is greatest, and running simulations on the fly to minimize it. The proposed workflow enables nanoconfinement effects to be rigorously considered at the mesoscale where complex connected sets of nanopores control key applications such as hydrocarbon recovery and CO2 sequestration.