 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA New Deep State-Space Analysis Framework for Patient Latent State Estimation and Classification from EHR Time Series Data
Jul 21, 2023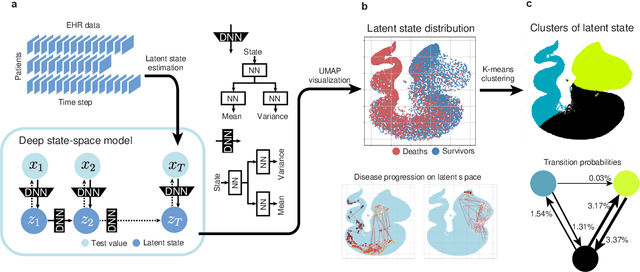
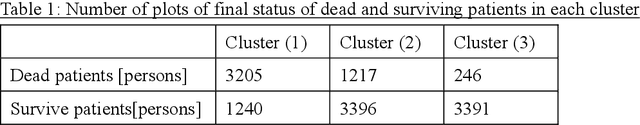
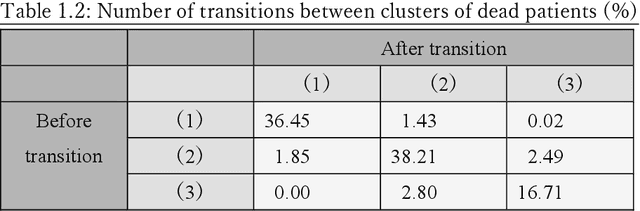
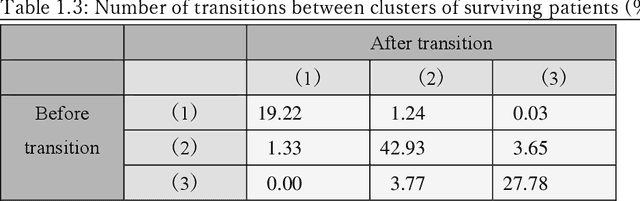
Many diseases, including cancer and chronic conditions, require extended treatment periods and long-term strategies. Machine learning and AI research focusing on electronic health records (EHRs) have emerged to address this need. Effective treatment strategies involve more than capturing sequential changes in patient test values. It requires an explainable and clinically interpretable model by capturing the patient's internal state over time. In this study, we propose the "deep state-space analysis framework," using time-series unsupervised learning of EHRs with a deep state-space model. This framework enables learning, visualizing, and clustering of temporal changes in patient latent states related to disease progression. We evaluated our framework using time-series laboratory data from 12,695 cancer patients. By estimating latent states, we successfully discover latent states related to prognosis. By visualization and cluster analysis, the temporal transition of patient status and test items during state transitions characteristic of each anticancer drug were identified. Our framework surpasses existing methods in capturing interpretable latent space. It can be expected to enhance our comprehension of disease progression from EHRs, aiding treatment adjustments and prognostic determinations.
An end-to-end framework for gene expression classification by integrating a background knowledge graph: application to cancer prognosis prediction
Jun 29, 2023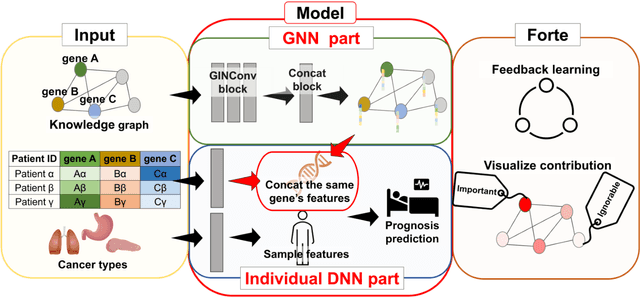
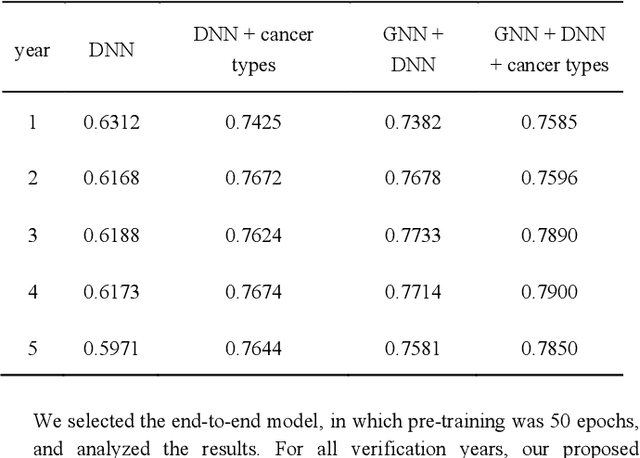
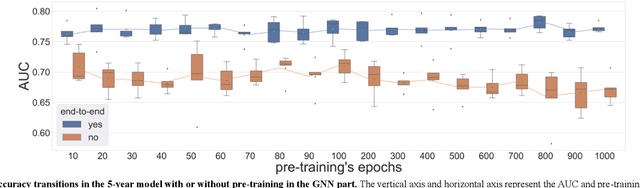
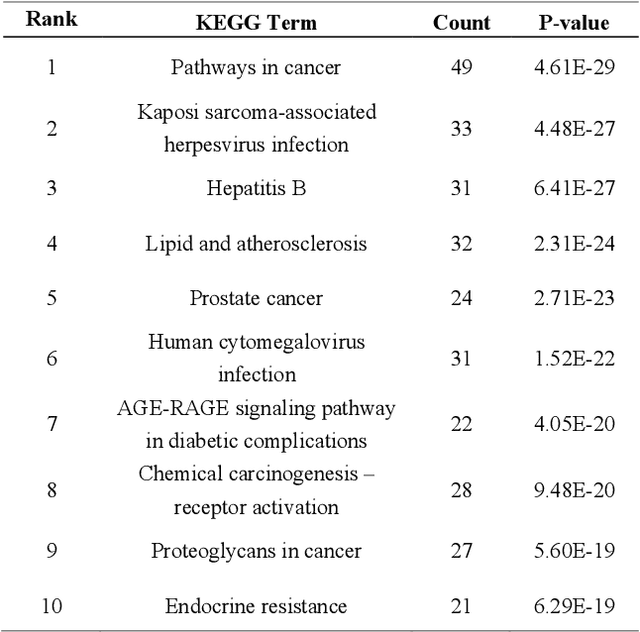
Biological data may be separated into primary data, such as gene expression, and secondary data, such as pathways and protein-protein interactions. Methods using secondary data to enhance the analysis of primary data are promising, because secondary data have background information that is not included in primary data. In this study, we proposed an end-to-end framework to integrally handle secondary data to construct a classification model for primary data. We applied this framework to cancer prognosis prediction using gene expression data and a biological network. Cross-validation results indicated that our model achieved higher accuracy compared with a deep neural network model without background biological network information. Experiments conducted in patient groups by cancer type showed improvement in ROC-area under the curve for many groups. Visualizations of high accuracy cancer types identified contributing genes and pathways by enrichment analysis. Known biomarkers and novel biomarker candidates were identified through these experiments.
GraphIX: Graph-based In silico XAI for drug repositioning from biopharmaceutical network
Dec 27, 2022Drug repositioning holds great promise because it can reduce the time and cost of new drug development. While drug repositioning can omit various R&D processes, confirming pharmacological effects on biomolecules is essential for application to new diseases. Biomedical explainability in a drug repositioning model can support appropriate insights in subsequent in-depth studies. However, the validity of the XAI methodology is still under debate, and the effectiveness of XAI in drug repositioning prediction applications remains unclear. In this study, we propose GraphIX, an explainable drug repositioning framework using biological networks, and quantitatively evaluate its explainability. GraphIX first learns the network weights and node features using a graph neural network from known drug indication and knowledge graph that consists of three types of nodes (but not given node type information): disease, drug, and protein. Analysis of the post-learning features showed that node types that were not known to the model beforehand are distinguished through the learning process based on the graph structure. From the learned weights and features, GraphIX then predicts the disease-drug association and calculates the contribution values of the nodes located in the neighborhood of the predicted disease and drug. We hypothesized that the neighboring protein node to which the model gave a high contribution is important in understanding the actual pharmacological effects. Quantitative evaluation of the validity of protein nodes' contribution using a real-world database showed that the high contribution proteins shown by GraphIX are reasonable as a mechanism of drug action. GraphIX is a framework for evidence-based drug discovery that can present to users new disease-drug associations and identify the protein important for understanding its pharmacological effects from a large and complex knowledge base.