 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAddressing the Scarcity of Benchmarks for Graph XAI
May 18, 2025While Graph Neural Networks (GNNs) have become the de facto model for learning from structured data, their decisional process remains opaque to the end user, undermining their deployment in safety-critical applications. In the case of graph classification, Explainable Artificial Intelligence (XAI) techniques address this major issue by identifying sub-graph motifs that explain predictions. However, advancements in this field are hindered by a chronic scarcity of benchmark datasets with known ground-truth motifs to assess the explanations' quality. Current graph XAI benchmarks are limited to synthetic data or a handful of real-world tasks hand-curated by domain experts. In this paper, we propose a general method to automate the construction of XAI benchmarks for graph classification from real-world datasets. We provide both 15 ready-made benchmarks, as well as the code to generate more than 2000 additional XAI benchmarks with our method. As a use case, we employ our benchmarks to assess the effectiveness of some popular graph explainers.
Learning to quantify graph nodes
Mar 19, 2025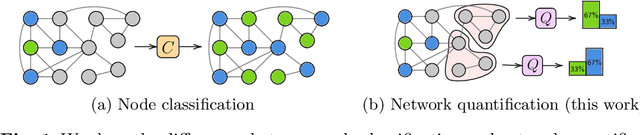
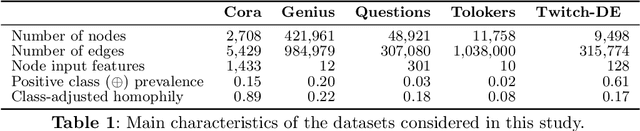
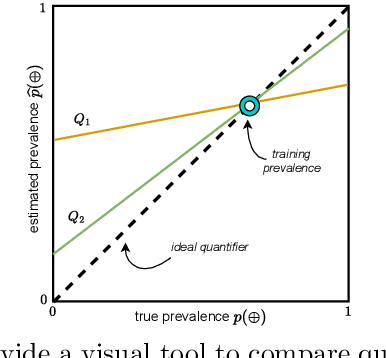
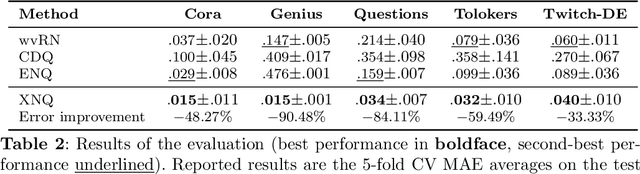
Network Quantification is the problem of estimating the class proportions in unlabeled subsets of graph nodes. When prior probability shift is at play, this task cannot be effectively addressed by first classifying the nodes and then counting the class predictions. In addition, unlike non-relational quantification on i.i.d. datapoints, Network Quantification demands enhanced flexibility to capture a broad range of connectivity patterns, resilience to the challenge of heterophily, and efficiency to scale to larger networks. To meet these stringent requirements we introduce XNQ, a novel method that synergizes the flexibility and efficiency of the unsupervised node embeddings computed by randomized recursive Graph Neural Networks, with an Expectation-Maximization algorithm that provides a robust quantification-aware adjustment to the output probabilities of a calibrated node classifier. We validate the design choices underpinning our method through comprehensive ablation experiments. In an extensive evaluation, we find that our approach consistently and significantly improves on the best Network Quantification methods to date, thereby setting the new state of the art for this challenging task. Simultaneously, it provides a training speed-up of up to 10x-100x over other graph learning based methods.
Towards Efficient Molecular Property Optimization with Graph Energy Based Models
Feb 17, 2025



Optimizing chemical properties is a challenging task due to the vastness and complexity of chemical space. Here, we present a generative energy-based architecture for implicit chemical property optimization, designed to efficiently generate molecules that satisfy target properties without explicit conditional generation. We use Graph Energy Based Models and a training approach that does not require property labels. We validated our approach on well-established chemical benchmarks, showing superior results to state-of-the-art methods and demonstrating robustness and efficiency towards de novo drug design.
Classifier-free graph diffusion for molecular property targeting
Dec 28, 2023
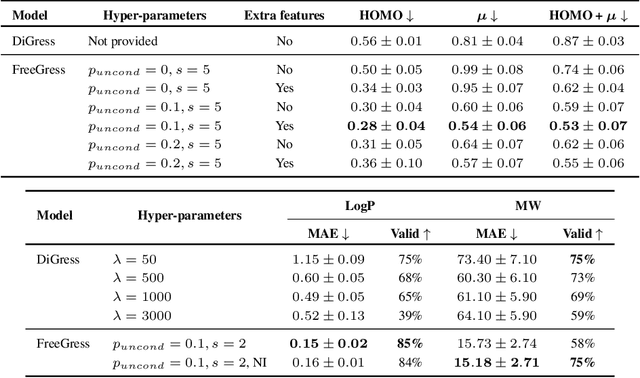
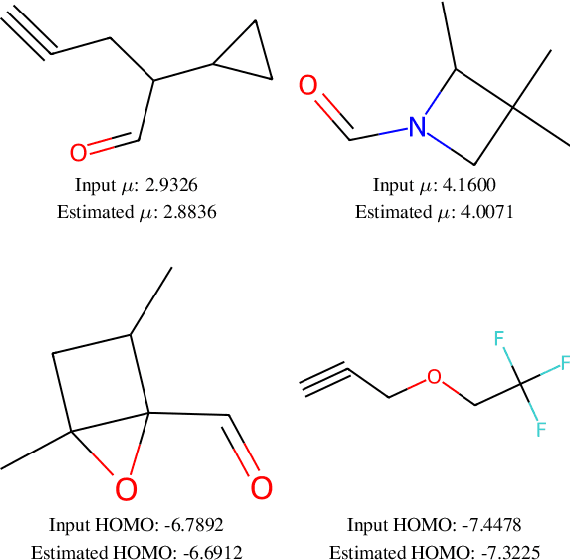
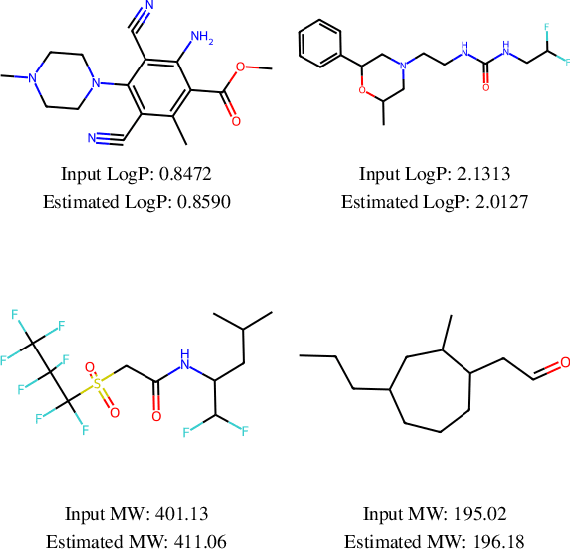
This work focuses on the task of property targeting: that is, generating molecules conditioned on target chemical properties to expedite candidate screening for novel drug and materials development. DiGress is a recent diffusion model for molecular graphs whose distinctive feature is allowing property targeting through classifier-based (CB) guidance. While CB guidance may work to generate molecular-like graphs, we hint at the fact that its assumptions apply poorly to the chemical domain. Based on this insight we propose a classifier-free DiGress (FreeGress), which works by directly injecting the conditioning information into the training process. CF guidance is convenient given its less stringent assumptions and since it does not require to train an auxiliary property regressor, thus halving the number of trainable parameters in the model. We empirically show that our model yields up to 79% improvement in Mean Absolute Error with respect to DiGress on property targeting tasks on QM9 and ZINC-250k benchmarks. As an additional contribution, we propose a simple yet powerful approach to improve chemical validity of generated samples, based on the observation that certain chemical properties such as molecular weight correlate with the number of atoms in molecules.
GraphGen-Redux: a Fast and Lightweight Recurrent Model for labeled Graph Generation
Jul 18, 2021
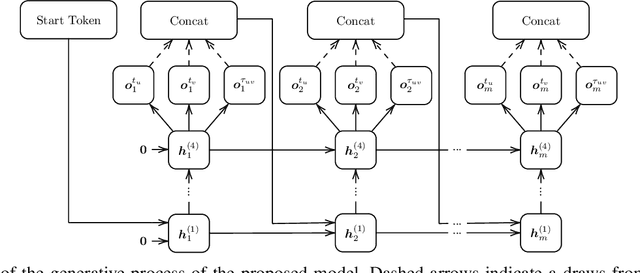
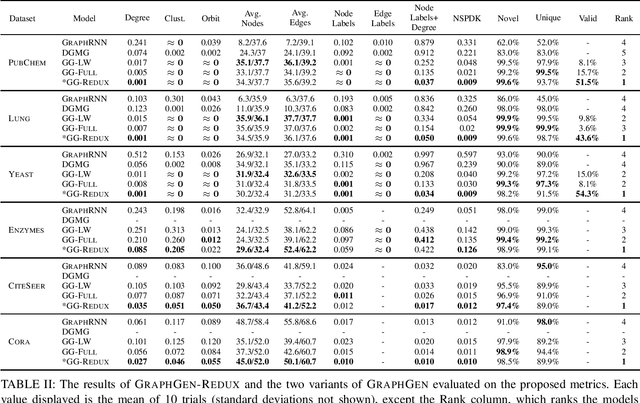
The problem of labeled graph generation is gaining attention in the Deep Learning community. The task is challenging due to the sparse and discrete nature of graph spaces. Several approaches have been proposed in the literature, most of which require to transform the graphs into sequences that encode their structure and labels and to learn the distribution of such sequences through an auto-regressive generative model. Among this family of approaches, we focus on the GraphGen model. The preprocessing phase of GraphGen transforms graphs into unique edge sequences called Depth-First Search (DFS) codes, such that two isomorphic graphs are assigned the same DFS code. Each element of a DFS code is associated with a graph edge: specifically, it is a quintuple comprising one node identifier for each of the two endpoints, their node labels, and the edge label. GraphGen learns to generate such sequences auto-regressively and models the probability of each component of the quintuple independently. While effective, the independence assumption made by the model is too loose to capture the complex label dependencies of real-world graphs precisely. By introducing a novel graph preprocessing approach, we are able to process the labeling information of both nodes and edges jointly. The corresponding model, which we term GraphGen-Redux, improves upon the generative performances of GraphGen in a wide range of datasets of chemical and social graphs. In addition, it uses approximately 78% fewer parameters than the vanilla variant and requires 50% fewer epochs of training on average.
A Deep Generative Model for Fragment-Based Molecule Generation
Feb 28, 2020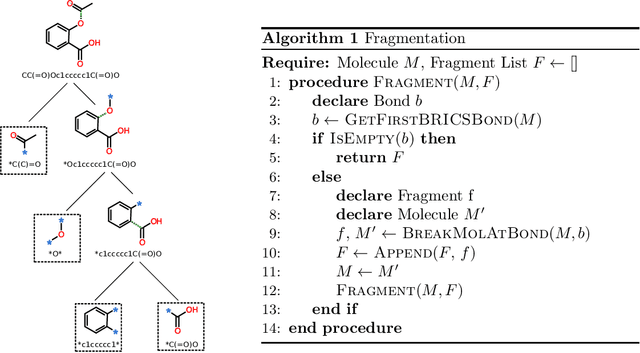
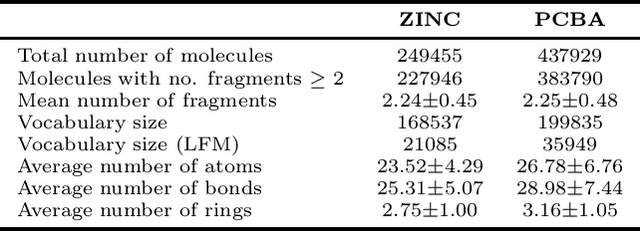
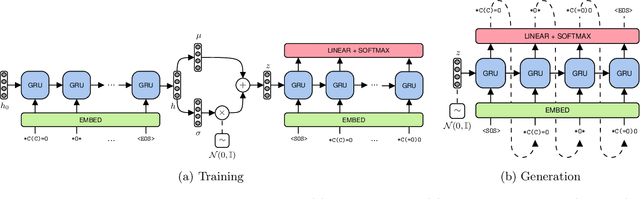
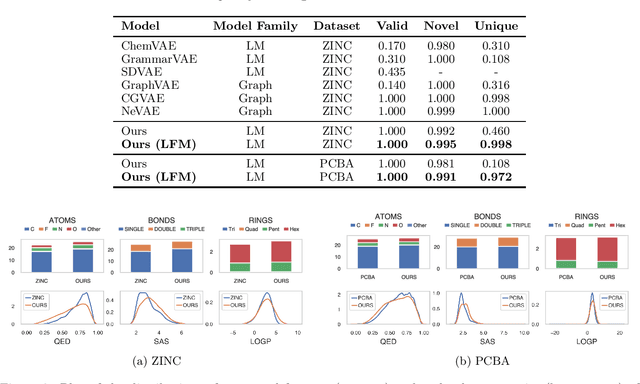
Molecule generation is a challenging open problem in cheminformatics. Currently, deep generative approaches addressing the challenge belong to two broad categories, differing in how molecules are represented. One approach encodes molecular graphs as strings of text, and learns their corresponding character-based language model. Another, more expressive, approach operates directly on the molecular graph. In this work, we address two limitations of the former: generation of invalid and duplicate molecules. To improve validity rates, we develop a language model for small molecular substructures called fragments, loosely inspired by the well-known paradigm of Fragment-Based Drug Design. In other words, we generate molecules fragment by fragment, instead of atom by atom. To improve uniqueness rates, we present a frequency-based masking strategy that helps generate molecules with infrequent fragments. We show experimentally that our model largely outperforms other language model-based competitors, reaching state-of-the-art performances typical of graph-based approaches. Moreover, generated molecules display molecular properties similar to those in the training sample, even in absence of explicit task-specific supervision.
Edge-based sequential graph generation with recurrent neural networks
Jan 31, 2020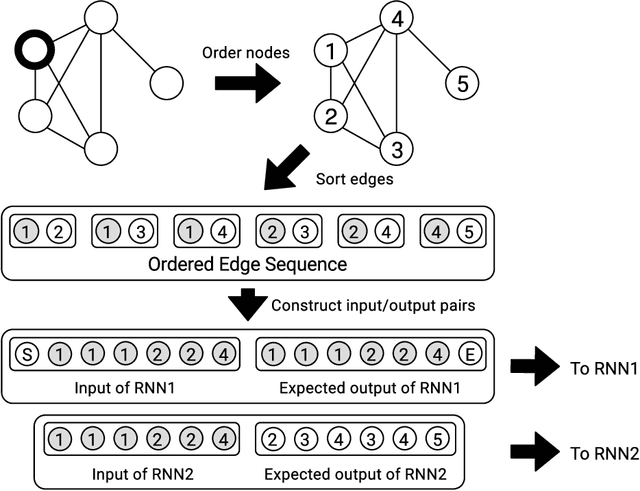
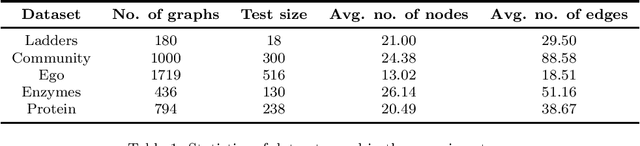
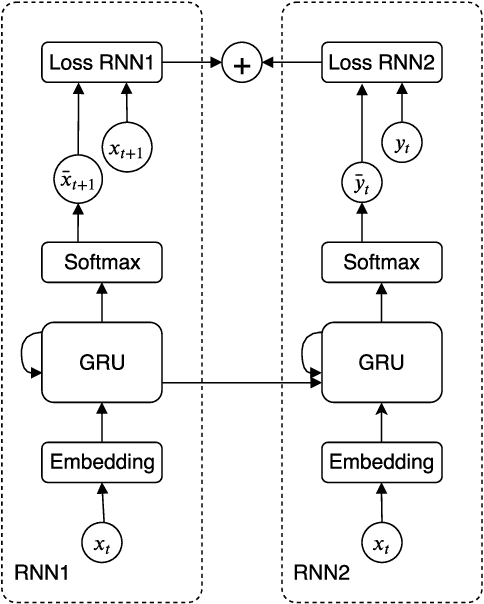
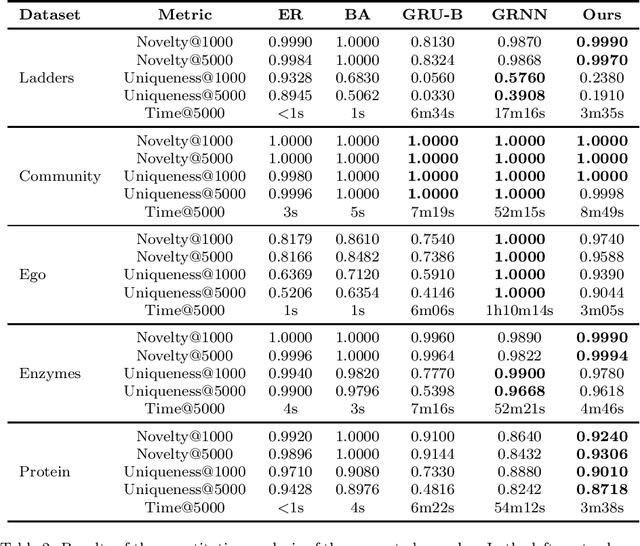
Graph generation with Machine Learning is an open problem with applications in various research fields. In this work, we propose to cast the generative process of a graph into a sequential one, relying on a node ordering procedure. We use this sequential process to design a novel generative model composed of two recurrent neural networks that learn to predict the edges of graphs: the first network generates one endpoint of each edge, while the second network generates the other endpoint conditioned on the state of the first. We test our approach extensively on five different datasets, comparing with two well-known baselines coming from graph literature, and two recurrent approaches, one of which holds state of the art performances. Evaluation is conducted considering quantitative and qualitative characteristics of the generated samples. Results show that our approach is able to yield novel, and unique graphs originating from very different distributions, while retaining structural properties very similar to those in the training sample. Under the proposed evaluation framework, our approach is able to reach performances comparable to the current state of the art on the graph generation task.
A Fair Comparison of Graph Neural Networks for Graph Classification
Jan 07, 2020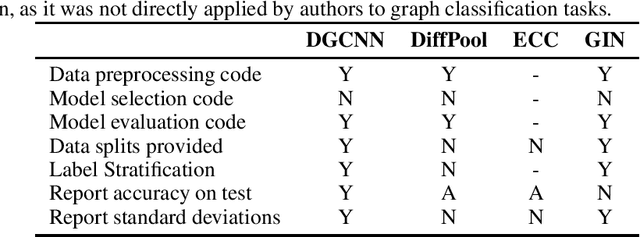
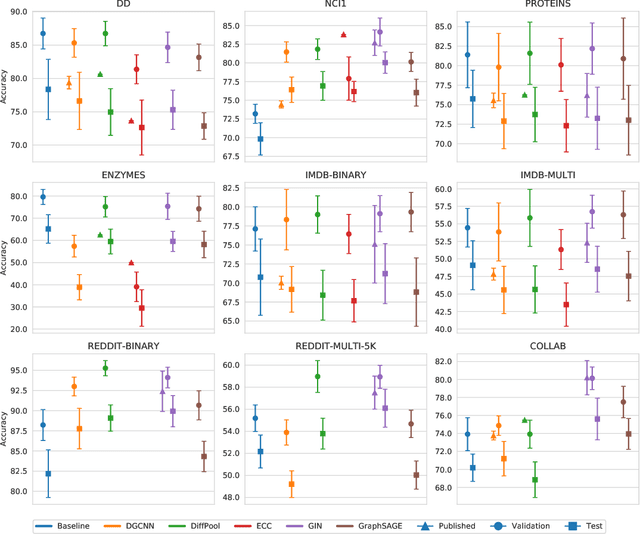

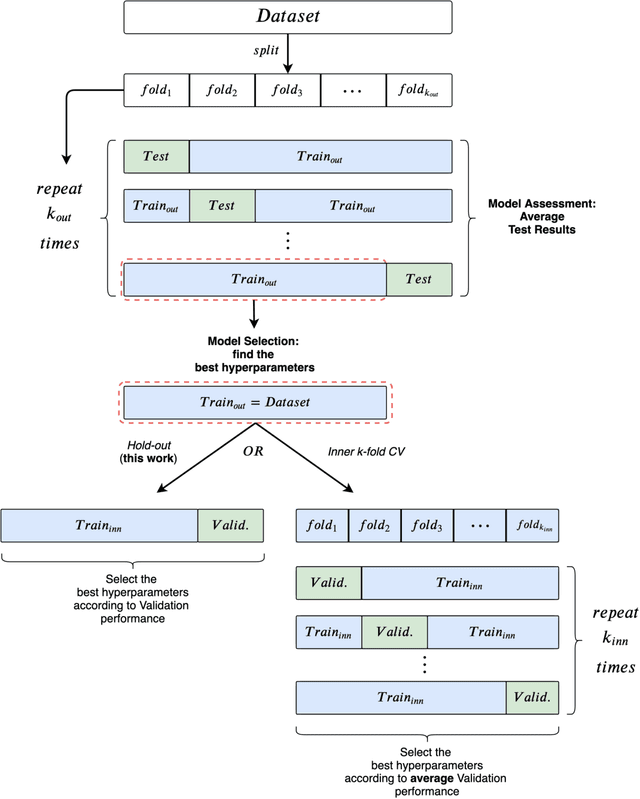
Experimental reproducibility and replicability are critical topics in machine learning. Authors have often raised concerns about their lack in scientific publications to improve the quality of the field. Recently, the graph representation learning field has attracted the attention of a wide research community, which resulted in a large stream of works. As such, several Graph Neural Network models have been developed to effectively tackle graph classification. However, experimental procedures often lack rigorousness and are hardly reproducible. Motivated by this, we provide an overview of common practices that should be avoided to fairly compare with the state of the art. To counter this troubling trend, we ran more than 47000 experiments in a controlled and uniform framework to re-evaluate five popular models across nine common benchmarks. Moreover, by comparing GNNs with structure-agnostic baselines we provide convincing evidence that, on some datasets, structural information has not been exploited yet. We believe that this work can contribute to the development of the graph learning field, by providing a much needed grounding for rigorous evaluations of graph classification models.
A Gentle Introduction to Deep Learning for Graphs
Dec 29, 2019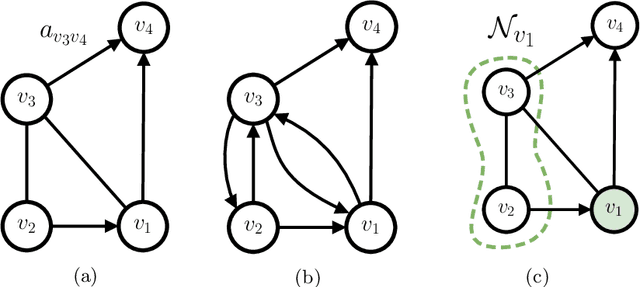

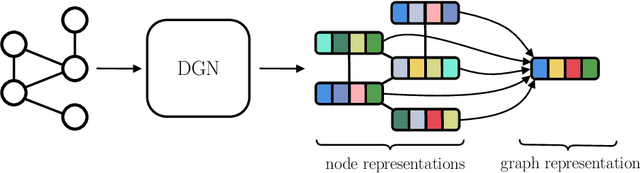

The adaptive processing of graph data is a long-standing research topic which has been lately consolidated as a theme of major interest in the deep learning community. The snap increase in the amount and breadth of related research has come at the price of little systematization of knowledge and attention to earlier literature. This work is designed as a tutorial introduction to the field of deep learning for graphs. It favours a consistent and progressive introduction of the main concepts and architectural aspects over an exposition of the most recent literature, for which the reader is referred to available surveys. The paper takes a top-down view to the problem, introducing a generalized formulation of graph representation learning based on a local and iterative approach to structured information processing. It introduces the basic building blocks that can be combined to design novel and effective neural models for graphs. The methodological exposition is complemented by a discussion of interesting research challenges and applications in the field.