 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSelf-Supervised Evolution Operator Learning for High-Dimensional Dynamical Systems
May 24, 2025
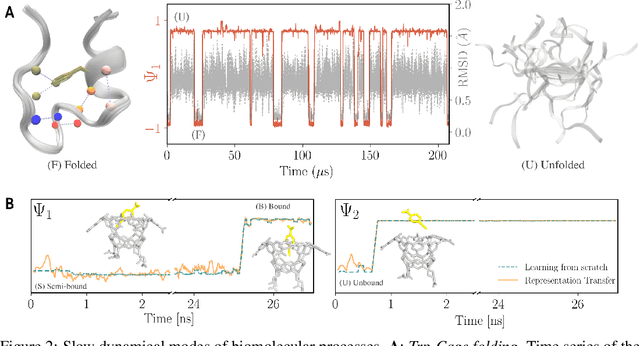
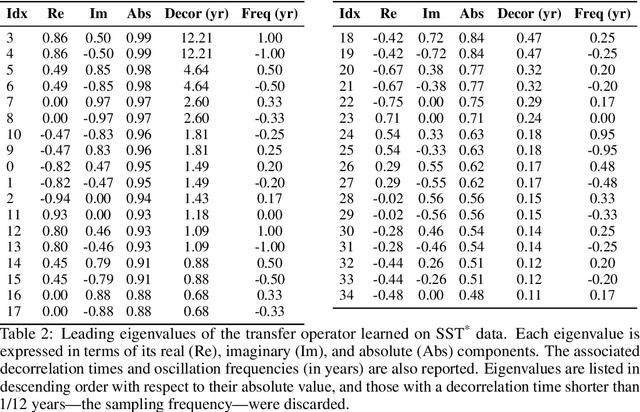
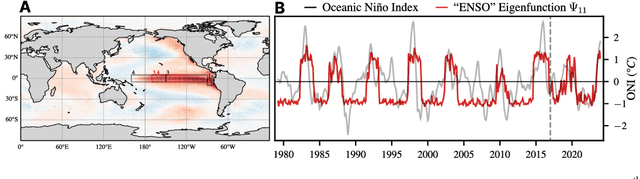
We introduce an encoder-only approach to learn the evolution operators of large-scale non-linear dynamical systems, such as those describing complex natural phenomena. Evolution operators are particularly well-suited for analyzing systems that exhibit complex spatio-temporal patterns and have become a key analytical tool across various scientific communities. As terabyte-scale weather datasets and simulation tools capable of running millions of molecular dynamics steps per day are becoming commodities, our approach provides an effective tool to make sense of them from a data-driven perspective. The core of it lies in a remarkable connection between self-supervised representation learning methods and the recently established learning theory of evolution operators. To show the usefulness of the proposed method, we test it across multiple scientific domains: explaining the folding dynamics of small proteins, the binding process of drug-like molecules in host sites, and autonomously finding patterns in climate data. Code and data to reproduce the experiments are made available open source.
Transfer learning for atomistic simulations using GNNs and kernel mean embeddings
Jun 02, 2023

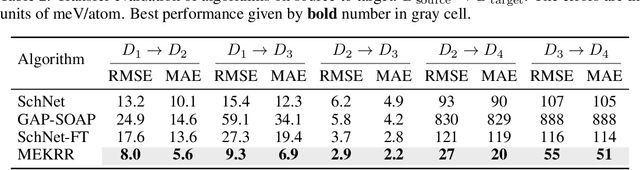

Interatomic potentials learned using machine learning methods have been successfully applied to atomistic simulations. However, deep learning pipelines are notoriously data-hungry, while generating reference calculations is computationally demanding. To overcome this difficulty, we propose a transfer learning algorithm that leverages the ability of graph neural networks (GNNs) in describing chemical environments, together with kernel mean embeddings. We extract a feature map from GNNs pre-trained on the OC20 dataset and use it to learn the potential energy surface from system-specific datasets of catalytic processes. Our method is further enhanced by a flexible kernel function that incorporates chemical species information, resulting in improved performance and interpretability. We test our approach on a series of realistic datasets of increasing complexity, showing excellent generalization and transferability performance, and improving on methods that rely on GNNs or ridge regression alone, as well as similar fine-tuning approaches. We make the code available to the community at https://github.com/IsakFalk/atomistic_transfer_mekrr.
Characterizing metastable states with the help of machine learning
Apr 15, 2022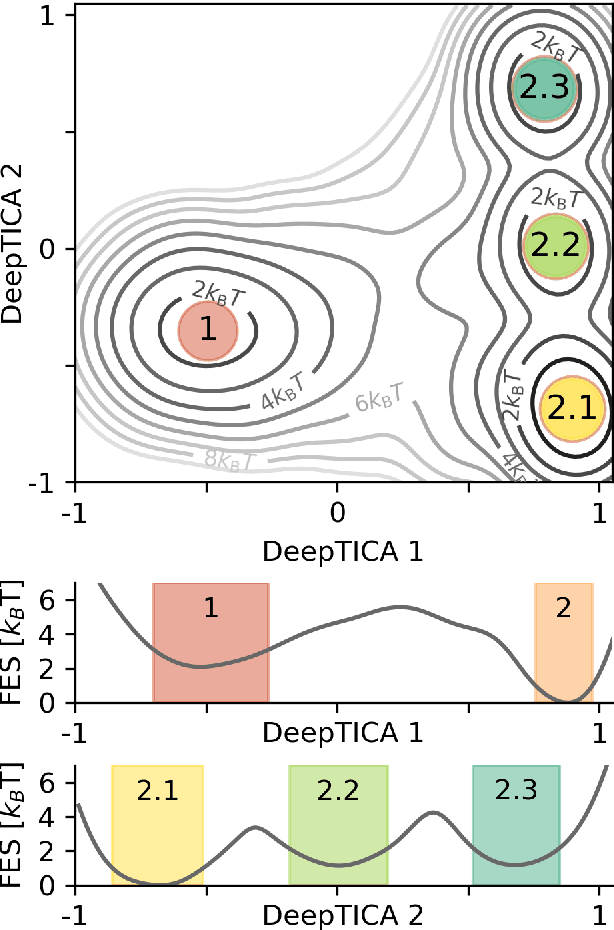
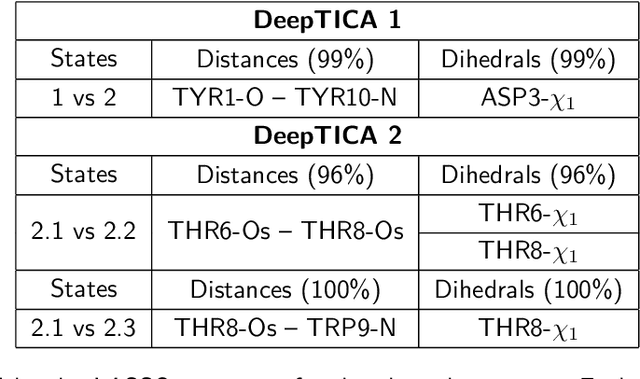


Present-day atomistic simulations generate long trajectories of ever more complex systems. Analyzing these data, discovering metastable states, and uncovering their nature is becoming increasingly challenging. In this paper, we first use the variational approach to conformation dynamics to discover the slowest dynamical modes of the simulations. This allows the different metastable states of the system to be located and organized hierarchically. The physical descriptors that characterize metastable states are discovered by means of a machine learning method. We show in the cases of two proteins, Chignolin and Bovine Pancreatic Trypsin Inhibitor, how such analysis can be effortlessly performed in a matter of seconds. Another strength of our approach is that it can be applied to the analysis of both unbiased and biased simulations.