 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIntegrating Chemical Language and Molecular Graph in Multimodal Fused Deep Learning for Drug Property Prediction
Dec 29, 2023



Accurately predicting molecular properties is a challenging but essential task in drug discovery. Recently, many mono-modal deep learning methods have been successfully applied to molecular property prediction. However, the inherent limitation of mono-modal learning arises from relying solely on one modality of molecular representation, which restricts a comprehensive understanding of drug molecules and hampers their resilience against data noise. To overcome the limitations, we construct multimodal deep learning models to cover different molecular representations. We convert drug molecules into three molecular representations, SMILES-encoded vectors, ECFP fingerprints, and molecular graphs. To process the modal information, Transformer-Encoder, bi-directional gated recurrent units (BiGRU), and graph convolutional network (GCN) are utilized for feature learning respectively, which can enhance the model capability to acquire complementary and naturally occurring bioinformatics information. We evaluated our triple-modal model on six molecule datasets. Different from bi-modal learning models, we adopt five fusion methods to capture the specific features and leverage the contribution of each modal information better. Compared with mono-modal models, our multimodal fused deep learning (MMFDL) models outperform single models in accuracy, reliability, and resistance capability against noise. Moreover, we demonstrate its generalization ability in the prediction of binding constants for protein-ligand complex molecules in the refined set of PDBbind. The advantage of the multimodal model lies in its ability to process diverse sources of data using proper models and suitable fusion methods, which would enhance the noise resistance of the model while obtaining data diversity.
DePS: An improved deep learning model for de novo peptide sequencing
Mar 16, 2022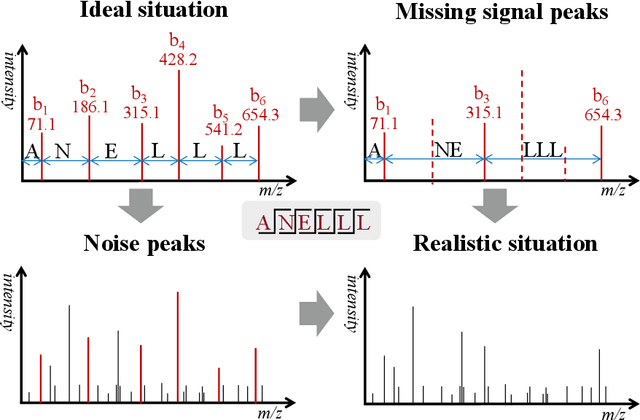
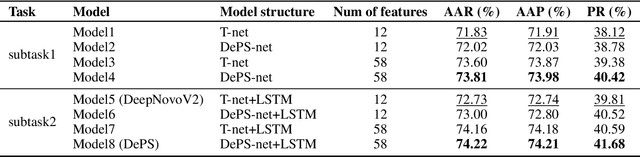
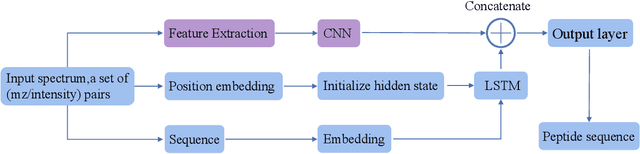
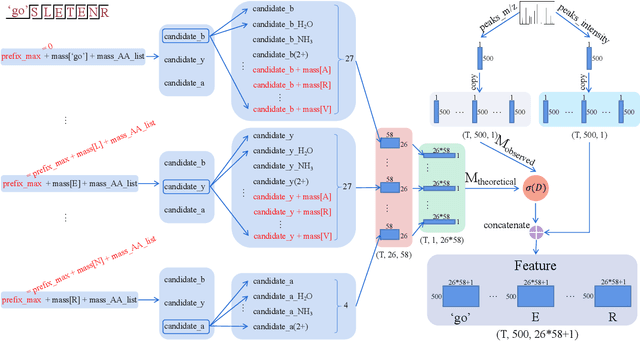
De novo peptide sequencing from mass spectrometry data is an important method for protein identification. Recently, various deep learning approaches were applied for de novo peptide sequencing and DeepNovoV2 is one of the represetative models. In this study, we proposed an enhanced model, DePS, which can improve the accuracy of de novo peptide sequencing even with missing signal peaks or large number of noisy peaks in tandem mass spectrometry data. It is showed that, for the same test set of DeepNovoV2, the DePS model achieved excellent results of 74.22%, 74.21% and 41.68% for amino acid recall, amino acid precision and peptide recall respectively. Furthermore, the results suggested that DePS outperforms DeepNovoV2 on the cross species dataset.