 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTransition Matrix Representation of Trees with Transposed Convolutions
Feb 22, 2022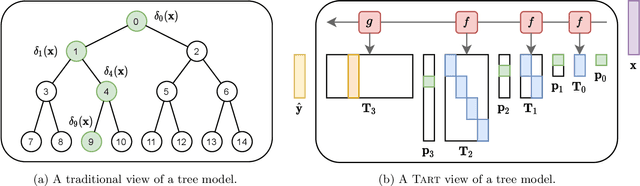
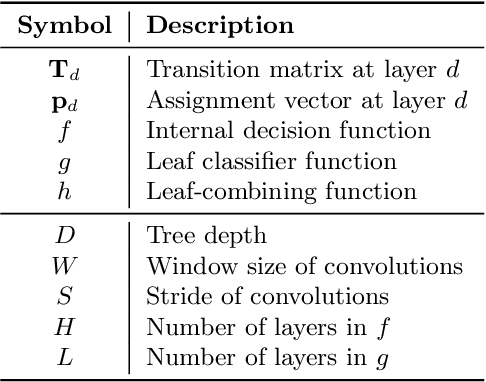
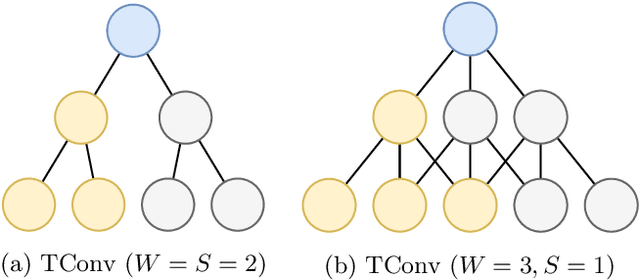

How can we effectively find the best structures in tree models? Tree models have been favored over complex black box models in domains where interpretability is crucial for making irreversible decisions. However, searching for a tree structure that gives the best balance between the performance and the interpretability remains a challenging task. In this paper, we propose TART (Transition Matrix Representation with Transposed Convolutions), our novel generalized tree representation for optimal structural search. TART represents a tree model with a series of transposed convolutions that boost the speed of inference by avoiding the creation of transition matrices. As a result, TART allows one to search for the best tree structure with a few design parameters, achieving higher classification accuracy than those of baseline models in feature-based datasets.
Fast and Accurate Pseudoinverse with Sparse Matrix Reordering and Incremental Approach
Nov 09, 2020
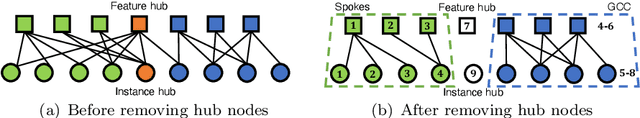

How can we compute the pseudoinverse of a sparse feature matrix efficiently and accurately for solving optimization problems? A pseudoinverse is a generalization of a matrix inverse, which has been extensively utilized as a fundamental building block for solving linear systems in machine learning. However, an approximate computation, let alone an exact computation, of pseudoinverse is very time-consuming due to its demanding time complexity, which limits it from being applied to large data. In this paper, we propose FastPI (Fast PseudoInverse), a novel incremental singular value decomposition (SVD) based pseudoinverse method for sparse matrices. Based on the observation that many real-world feature matrices are sparse and highly skewed, FastPI reorders and divides the feature matrix and incrementally computes low-rank SVD from the divided components. To show the efficacy of proposed FastPI, we apply them in real-world multi-label linear regression problems. Through extensive experiments, we demonstrate that FastPI computes the pseudoinverse faster than other approximate methods without loss of accuracy. %and uses much less memory compared to full-rank SVD based approach. Results imply that our method efficiently computes the low-rank pseudoinverse of a large and sparse matrix that other existing methods cannot handle with limited time and space.
SNeCT: Scalable network constrained Tucker decomposition for integrative multi-platform data analysis
Nov 26, 2017

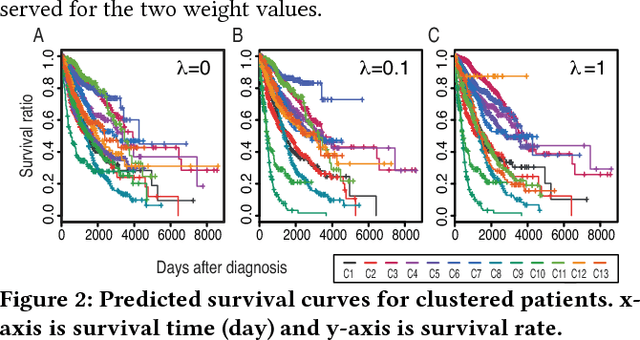
Motivation: How do we integratively analyze large-scale multi-platform genomic data that are high dimensional and sparse? Furthermore, how can we incorporate prior knowledge, such as the association between genes, in the analysis systematically? Method: To solve this problem, we propose a Scalable Network Constrained Tucker decomposition method we call SNeCT. SNeCT adopts parallel stochastic gradient descent approach on the proposed parallelizable network constrained optimization function. SNeCT decomposition is applied to tensor constructed from large scale multi-platform multi-cohort cancer data, PanCan12, constrained on a network built from PathwayCommons database. Results: The decomposed factor matrices are applied to stratify cancers, to search for top-k similar patients, and to illustrate how the matrices can be used for personalized interpretation. In the stratification test, combined twelve-cohort data is clustered to form thirteen subclasses. The thirteen subclasses have a high correlation to tissue of origin in addition to other interesting observations, such as clear separation of OV cancers to two groups, and high clinical correlation within subclusters formed in cohorts BRCA and UCEC. In the top-k search, a new patient's genomic profile is generated and searched against existing patients based on the factor matrices. The similarity of the top-k patient to the query is high for 23 clinical features, including estrogen/progesterone receptor statuses of BRCA patients with average precision value ranges from 0.72 to 0.86 and from 0.68 to 0.86, respectively. We also provide an illustration of how the factor matrices can be used for interpretable personalized analysis of each patient.
Feature versus Raw Sequence: Deep Learning Comparative Study on Predicting Pre-miRNA
Oct 17, 2017

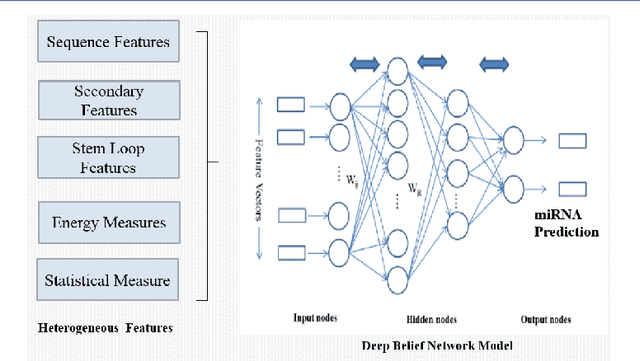

Should we input known genome sequence features or input sequence itself in deep learning framework? As deep learning more popular in various applications, researchers often come to question whether to generate features or use raw sequences for deep learning. To answer this question, we study the prediction accuracy of precursor miRNA prediction of feature-based deep belief network and sequence-based convolution neural network. Tested on a variant of six-layer convolution neural net and three-layer deep belief network, we find the raw sequence input based convolution neural network model performs similar or slightly better than feature based deep belief networks with best accuracy values of 0.995 and 0.990, respectively. Both the models outperform existing benchmarks models. The results shows us that if provided large enough data, well devised raw sequence based deep learning models can replace feature based deep learning models. However, construction of well behaved deep learning model can be very challenging. In cased features can be easily extracted, feature-based deep learning models may be a better alternative.
Multi-Kernel LS-SVM Based Bio-Clinical Data Integration: Applications to Ovarian Cancer
Oct 10, 2017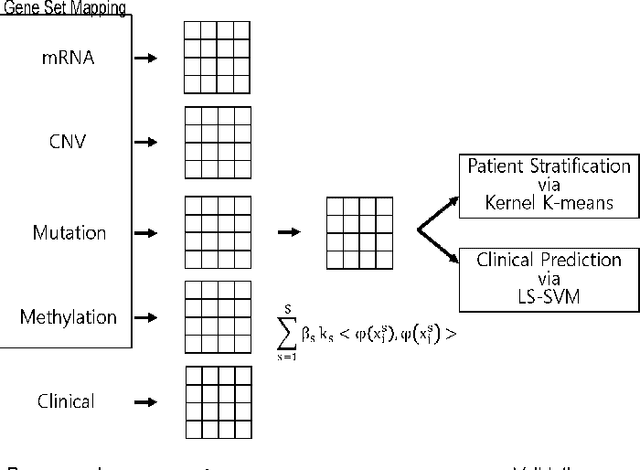
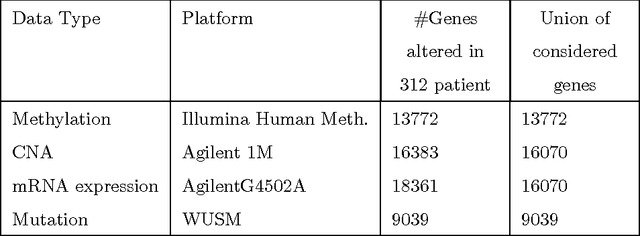
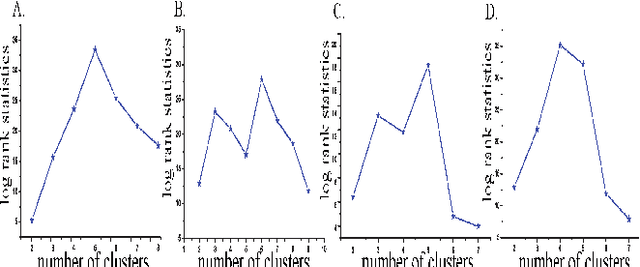
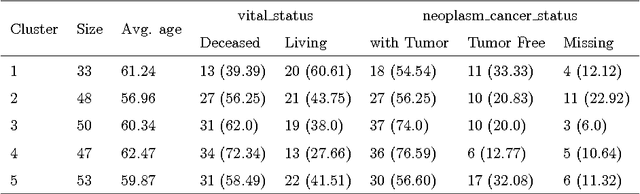
The medical research facilitates to acquire a diverse type of data from the same individual for particular cancer. Recent studies show that utilizing such diverse data results in more accurate predictions. The major challenge faced is how to utilize such diverse data sets in an effective way. In this paper, we introduce a multiple kernel based pipeline for integrative analysis of high-throughput molecular data (somatic mutation, copy number alteration, DNA methylation and mRNA) and clinical data. We apply the pipeline on Ovarian cancer data from TCGA. After multiple kernels have been generated from the weighted sum of individual kernels, it is used to stratify patients and predict clinical outcomes. We examine the survival time, vital status, and neoplasm cancer status of each subtype to verify how well they cluster. We have also examined the power of molecular and clinical data in predicting dichotomized overall survival data and to classify the tumor grade for the cancer samples. It was observed that the integration of various data types yields higher log-rank statistics value. We were also able to predict clinical status with higher accuracy as compared to using individual data types.
Prior Knowledge based mutation prioritization towards causal variant finding in rare disease
Oct 10, 2017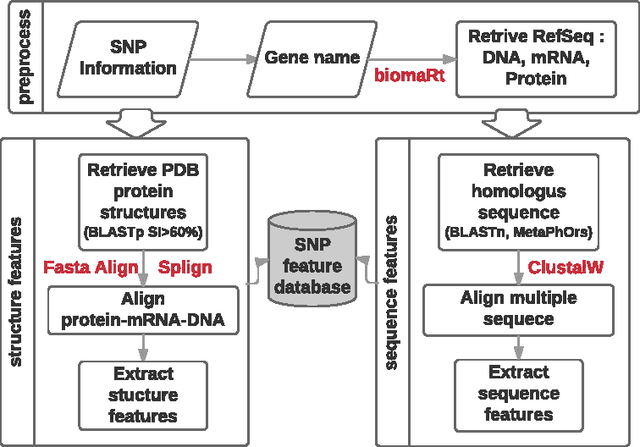
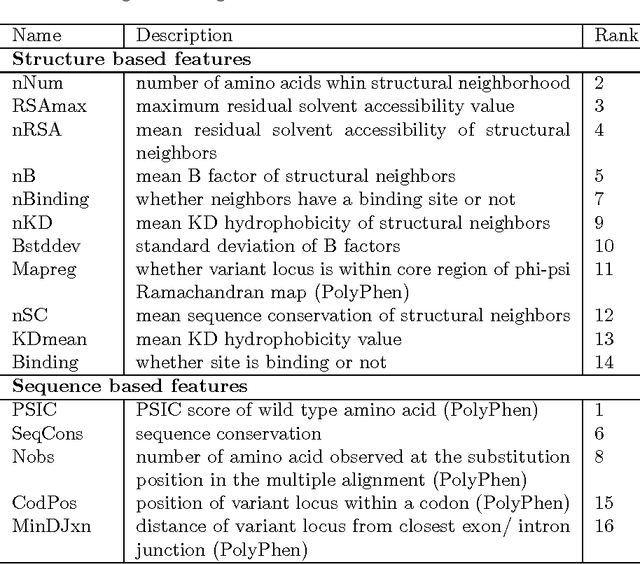
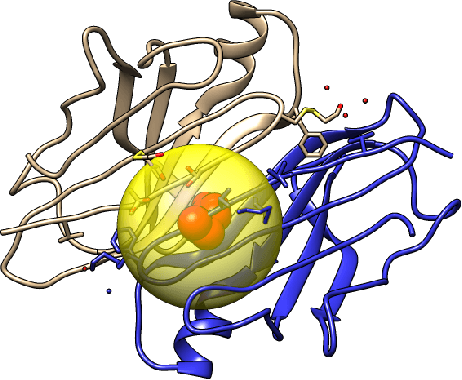
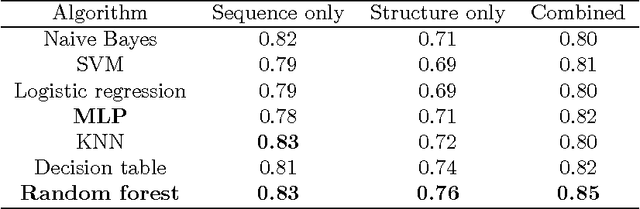
How do we determine the mutational effects in exome sequencing data with little or no statistical evidence? Can protein structural information fill in the gap of not having enough statistical evidence? In this work, we answer the two questions with the goal towards determining pathogenic effects of rare variants in rare disease. We take the approach of determining the importance of point mutation loci focusing on protein structure features. The proposed structure-based features contain information about geometric, physicochemical, and functional information of mutation loci and those of structural neighbors of the loci. The performance of the structure-based features trained on 80\% of HumDiv and tested on 20\% of HumDiv and on ClinVar datasets showed high levels of discernibility in the mutation's pathogenic or benign effects: F score of 0.71 and 0.68 respectively using multi-layer perceptron. Combining structure- and sequence-based feature further improve the accuracy: F score of 0.86 (HumDiv) and 0.75 (ClinVar). Also, careful examination of the rare variants in rare diseases cases showed that structure-based features are important in discerning importance of variant loci.
CTD: Fast, Accurate, and Interpretable Method for Static and Dynamic Tensor Decompositions
Oct 09, 2017
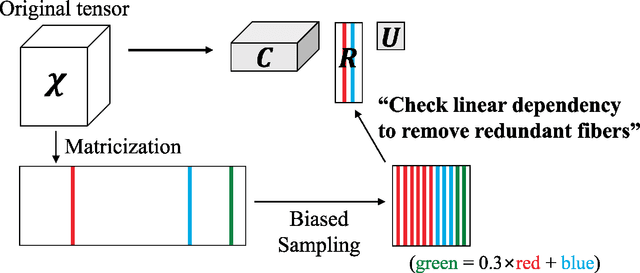

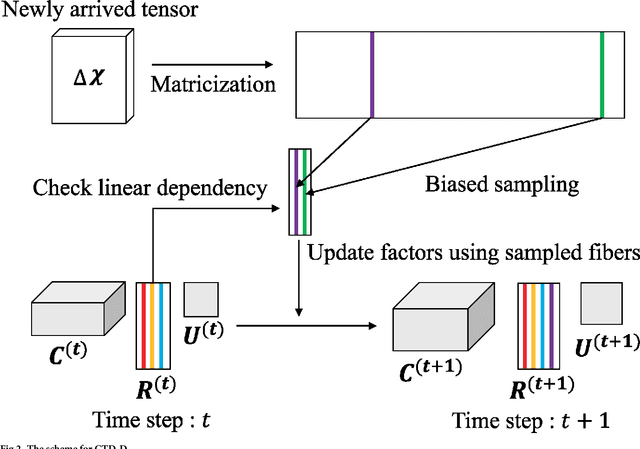
How can we find patterns and anomalies in a tensor, or multi-dimensional array, in an efficient and directly interpretable way? How can we do this in an online environment, where a new tensor arrives each time step? Finding patterns and anomalies in a tensor is a crucial problem with many applications, including building safety monitoring, patient health monitoring, cyber security, terrorist detection, and fake user detection in social networks. Standard PARAFAC and Tucker decomposition results are not directly interpretable. Although a few sampling-based methods have previously been proposed towards better interpretability, they need to be made faster, more memory efficient, and more accurate. In this paper, we propose CTD, a fast, accurate, and directly interpretable tensor decomposition method based on sampling. CTD-S, the static version of CTD, provably guarantees a high accuracy that is 17 ~ 83x more accurate than that of the state-of-the-art method. Also, CTD-S is made 5 ~ 86x faster, and 7 ~ 12x more memory-efficient than the state-of-the-art method by removing redundancy. CTD-D, the dynamic version of CTD, is the first interpretable dynamic tensor decomposition method ever proposed. Also, it is made 2 ~ 3x faster than already fast CTD-S by exploiting factors at previous time step and by reordering operations. With CTD, we demonstrate how the results can be effectively interpreted in the online distributed denial of service (DDoS) attack detection.
Next Generation Business Intelligence and Analytics: A Survey
Apr 11, 2017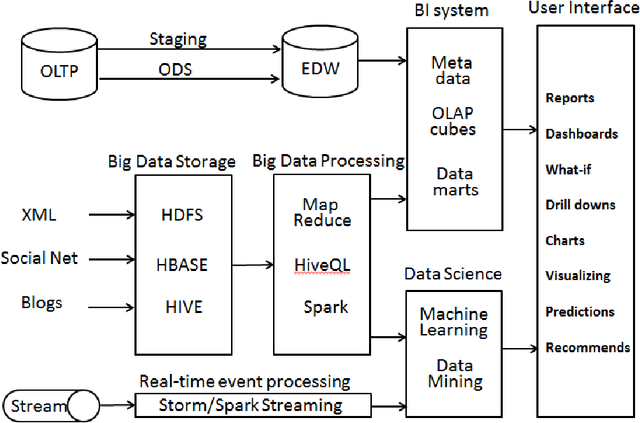
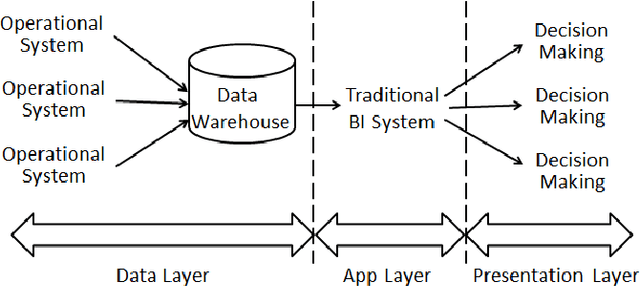
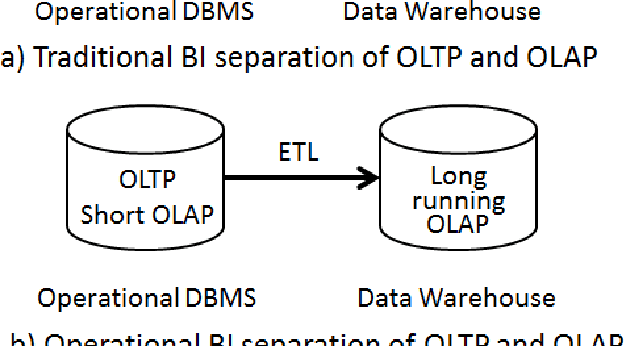
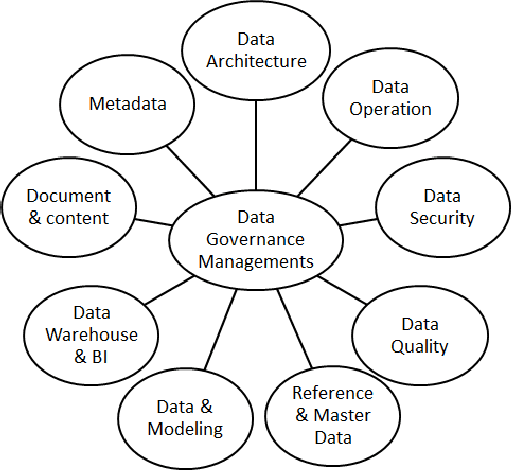
Business Intelligence and Analytics (BI&A) is the process of extracting and predicting business-critical insights from data. Traditional BI focused on data collection, extraction, and organization to enable efficient query processing for deriving insights from historical data. With the rise of big data and cloud computing, there are many challenges and opportunities for the BI. Especially with the growing number of data sources, traditional BI\&A are evolving to provide intelligence at different scales and perspectives - operational BI, situational BI, self-service BI. In this survey, we review the evolution of business intelligence systems in full scale from back-end architecture to and front-end applications. We focus on the changes in the back-end architecture that deals with the collection and organization of the data. We also review the changes in the front-end applications, where analytic services and visualization are the core components. Using a uses case from BI in Healthcare, which is one of the most complex enterprises, we show how BI\&A will play an important role beyond the traditional usage. The survey provides a holistic view of Business Intelligence and Analytics for anyone interested in getting a complete picture of the different pieces in the emerging next generation BI\&A solutions.
Deep Neural Network Based Precursor microRNA Prediction on Eleven Species
Apr 10, 2017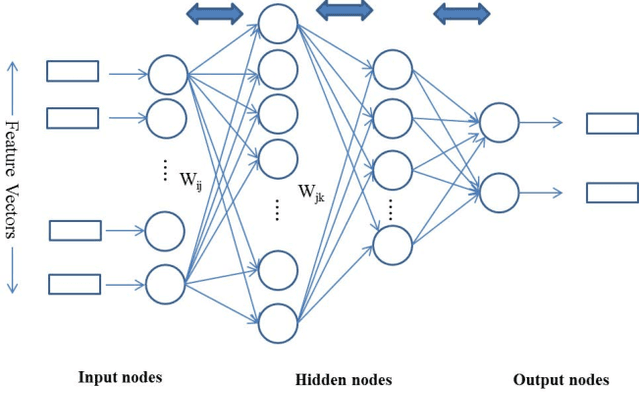
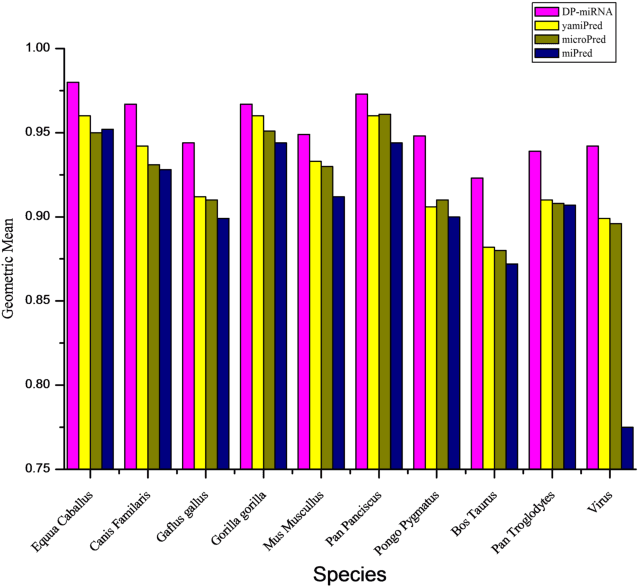
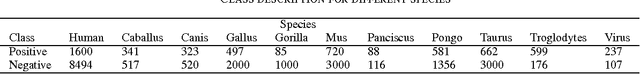
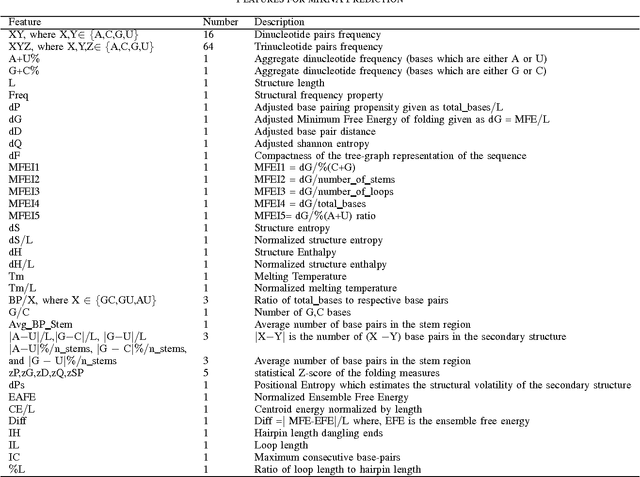
MicroRNA (miRNA) are small non-coding RNAs that regulates the gene expression at the post-transcriptional level. Determining whether a sequence segment is miRNA is experimentally challenging. Also, experimental results are sensitive to the experimental environment. These limitations inspire the development of computational methods for predicting the miRNAs. We propose a deep learning based classification model, called DP-miRNA, for predicting precursor miRNA sequence that contains the miRNA sequence. The feature set based Restricted Boltzmann Machine method, which we call DP-miRNA, uses 58 features that are categorized into four groups: sequence features, folding measures, stem-loop features and statistical feature. We evaluate the performance of the DP-miRNA on eleven twelve data sets of varying species, including the human. The deep neural network based classification outperformed support vector machine, neural network, naive Baye's classifiers, k-nearest neighbors, random forests, and a hybrid system combining support vector machine and genetic algorithm.