 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVon Mises Mixture Distributions for Molecular Conformation Generation
Jun 13, 2023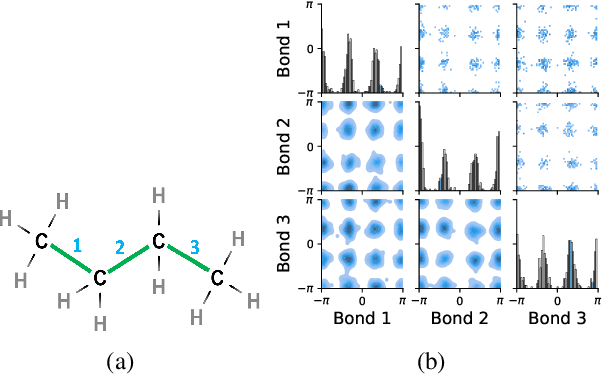
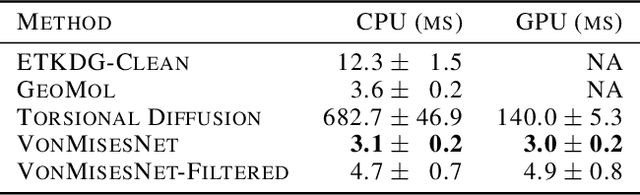
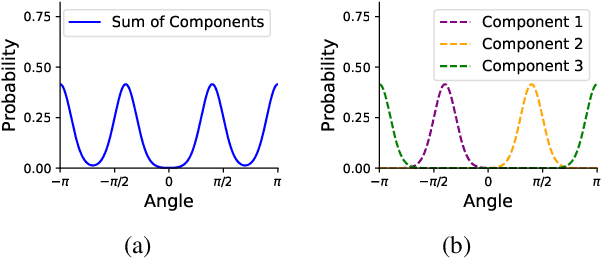
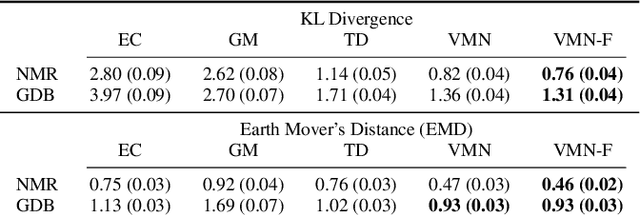
Molecules are frequently represented as graphs, but the underlying 3D molecular geometry (the locations of the atoms) ultimately determines most molecular properties. However, most molecules are not static and at room temperature adopt a wide variety of geometries or $\textit{conformations}$. The resulting distribution on geometries $p(x)$ is known as the Boltzmann distribution, and many molecular properties are expectations computed under this distribution. Generating accurate samples from the Boltzmann distribution is therefore essential for computing these expectations accurately. Traditional sampling-based methods are computationally expensive, and most recent machine learning-based methods have focused on identifying $\textit{modes}$ in this distribution rather than generating true $\textit{samples}$. Generating such samples requires capturing conformational variability, and it has been widely recognized that the majority of conformational variability in molecules arises from rotatable bonds. In this work, we present VonMisesNet, a new graph neural network that captures conformational variability via a variational approximation of rotatable bond torsion angles as a mixture of von Mises distributions. We demonstrate that VonMisesNet can generate conformations for arbitrary molecules in a way that is both physically accurate with respect to the Boltzmann distribution and orders of magnitude faster than existing sampling methods.
Deep Learning for Automated Classification and Characterization of Amorphous Materials
Sep 10, 2019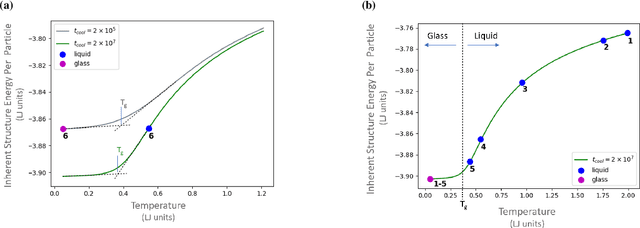
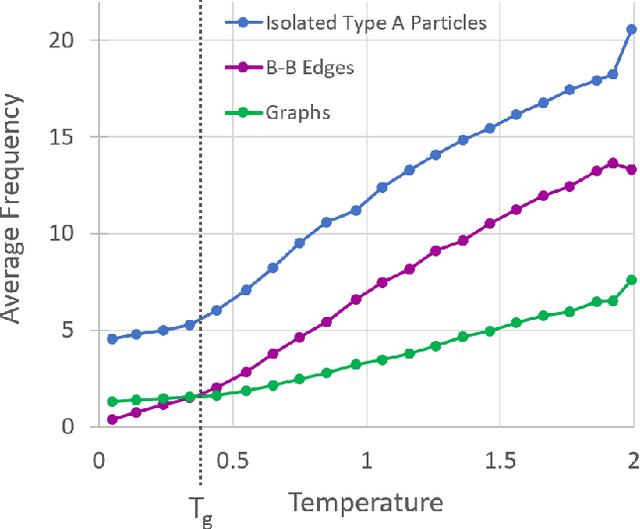
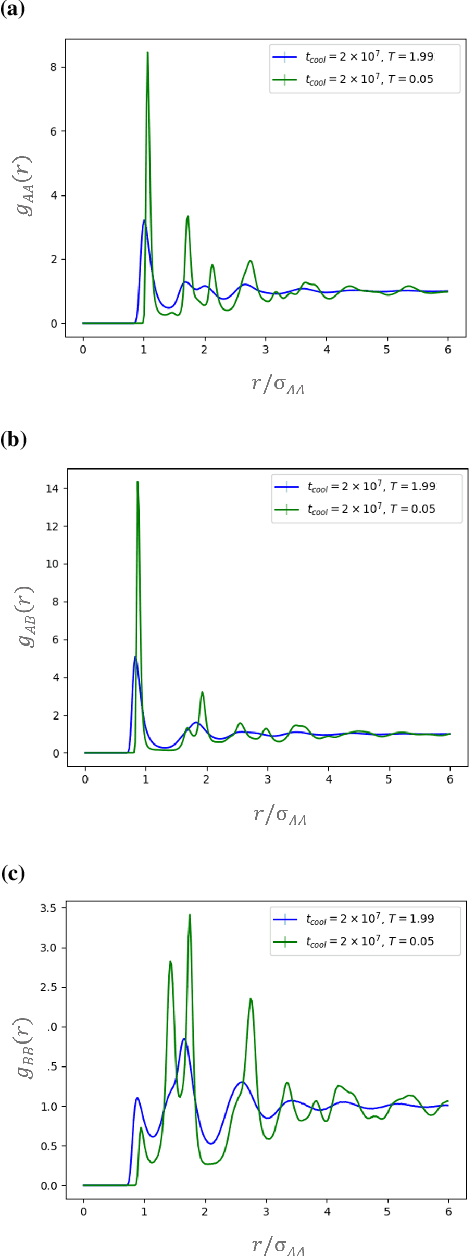
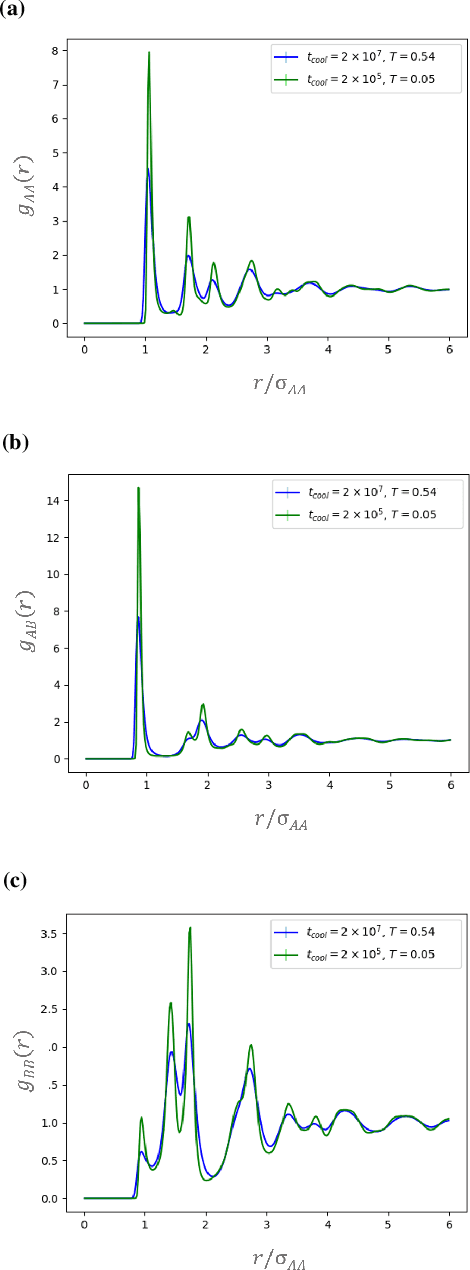
It is difficult to quantify structure-property relationships and to identify structural features of complex materials. The characterization of amorphous materials is especially challenging because their lack of long-range order makes it difficult to define structural metrics. In this work, we apply deep learning algorithms to accurately classify amorphous materials and characterize their structural features. Specifically, we show that convolutional neural networks and message passing neural networks can classify two-dimensional liquids and liquid-cooled glasses from molecular dynamics simulations with greater than 0.98 AUC, with no a priori assumptions about local particle relationships, even when the liquids and glasses are prepared at the same inherent structure energy. Furthermore, we demonstrate that message passing neural networks surpass convolutional neural networks in this context in both accuracy and interpretability. We extract a clear interpretation of how message passing neural networks evaluate liquid and glass structures by using a self-attention mechanism. Using this interpretation, we derive three novel structural metrics that accurately characterize glass formation. The methods presented here provide us with a procedure to identify important structural features in materials that could be missed by standard techniques and give us a unique insight into how these neural networks process data.