 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative AI-driven forecasting of oil production
Sep 24, 2024Forecasting oil production from oilfields with multiple wells is an important problem in petroleum and geothermal energy extraction, as well as energy storage technologies. The accuracy of oil forecasts is a critical determinant of economic projections, hydrocarbon reserves estimation, construction of fluid processing facilities, and energy price fluctuations. Leveraging generative AI techniques, we model time series forecasting of oil and water productions across four multi-well sites spanning four decades. Our goal is to effectively model uncertainties and make precise forecasts to inform decision-making processes at the field scale. We utilize an autoregressive model known as TimeGrad and a variant of a transformer architecture named Informer, tailored specifically for forecasting long sequence time series data. Predictions from both TimeGrad and Informer closely align with the ground truth data. The overall performance of the Informer stands out, demonstrating greater efficiency compared to TimeGrad in forecasting oil production rates across all sites.
Allegro-Legato: Scalable, Fast, and Robust Neural-Network Quantum Molecular Dynamics via Sharpness-Aware Minimization
Mar 14, 2023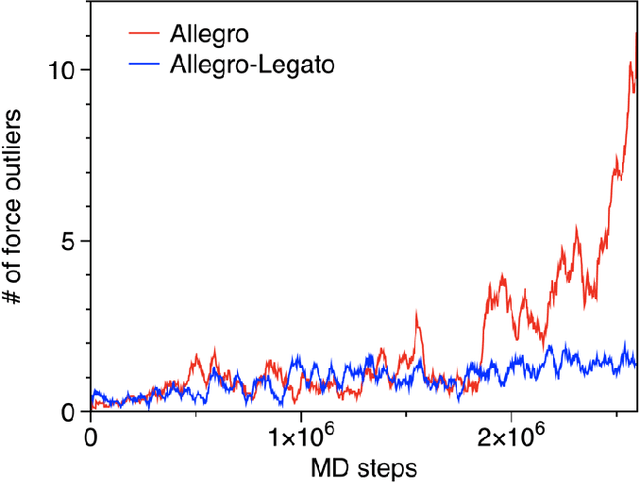

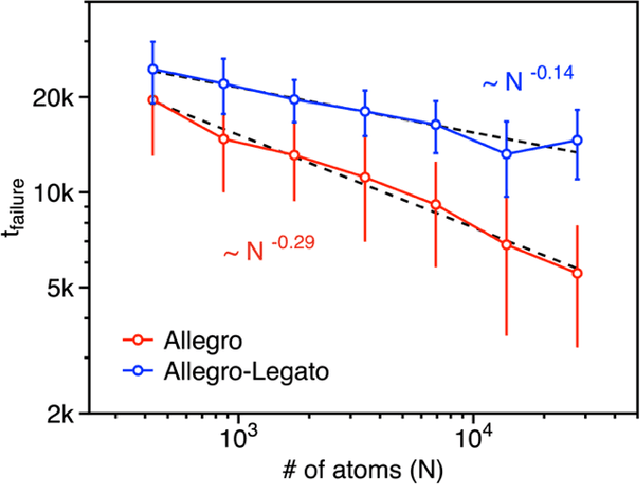

Neural-network quantum molecular dynamics (NNQMD) simulations based on machine learning are revolutionizing atomistic simulations of materials by providing quantum-mechanical accuracy but orders-of-magnitude faster, illustrated by ACM Gordon Bell prize (2020) and finalist (2021). State-of-the-art (SOTA) NNQMD model founded on group theory featuring rotational equivariance and local descriptors has provided much higher accuracy and speed than those models, thus named Allegro (meaning fast). On massively parallel supercomputers, however, it suffers a fidelity-scaling problem, where growing number of unphysical predictions of interatomic forces prohibits simulations involving larger numbers of atoms for longer times. Here, we solve this problem by combining the Allegro model with sharpness aware minimization (SAM) for enhancing the robustness of model through improved smoothness of the loss landscape. The resulting Allegro-Legato (meaning fast and "smooth") model was shown to elongate the time-to-failure $t_\textrm{failure}$, without sacrificing computational speed or accuracy. Specifically, Allegro-Legato exhibits much weaker dependence of timei-to-failure on the problem size, $t_{\textrm{failure}} \propto N^{-0.14}$ ($N$ is the number of atoms) compared to the SOTA Allegro model $\left(t_{\textrm{failure}} \propto N^{-0.29}\right)$, i.e., systematically delayed time-to-failure, thus allowing much larger and longer NNQMD simulations without failure. The model also exhibits excellent computational scalability and GPU acceleration on the Polaris supercomputer at Argonne Leadership Computing Facility. Such scalable, accurate, fast and robust NNQMD models will likely find broad applications in NNQMD simulations on emerging exaflop/s computers, with a specific example of accounting for nuclear quantum effects in the dynamics of ammonia.
Multiscale Graph Neural Networks for Protein Residue Contact Map Prediction
Dec 22, 2022



Machine learning (ML) is revolutionizing protein structural analysis, including an important subproblem of predicting protein residue contact maps, i.e., which amino-acid residues are in close spatial proximity given the amino-acid sequence of a protein. Despite recent progresses in ML-based protein contact prediction, predicting contacts with a wide range of distances (commonly classified into short-, medium- and long-range contacts) remains a challenge. Here, we propose a multiscale graph neural network (GNN) based approach taking a cue from multiscale physics simulations, in which a standard pipeline involving a recurrent neural network (RNN) is augmented with three GNNs to refine predictive capability for short-, medium- and long-range residue contacts, respectively. Test results on the ProteinNet dataset show improved accuracy for contacts of all ranges using the proposed multiscale RNN+GNN approach over the conventional approach, including the most challenging case of long-range contact prediction.
Physics-informed Neural-Network Software for Molecular Dynamics Applications
Nov 11, 2020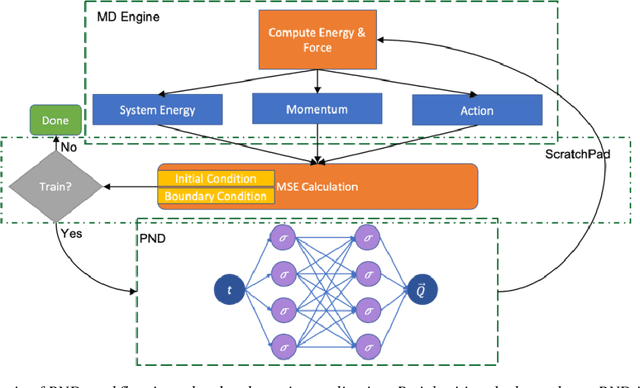

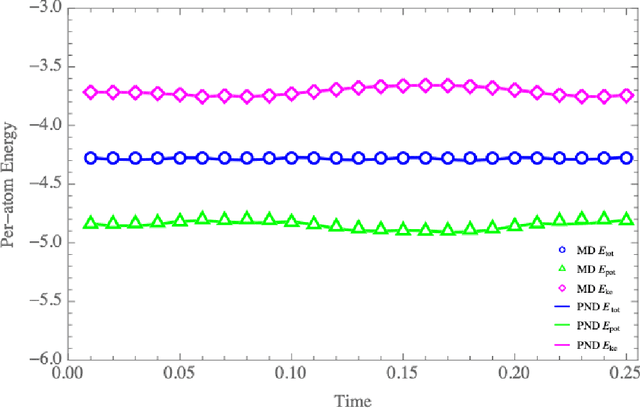
We have developed a novel differential equation solver software called PND based on the physics-informed neural network for molecular dynamics simulators. Based on automatic differentiation technique provided by Pytorch, our software allows users to flexibly implement equation of atom motions, initial and boundary conditions, and conservation laws as loss function to train the network. PND comes with a parallel molecular dynamics (MD) engine in order for users to examine and optimize loss function design, and different conservation laws and boundary conditions, and hyperparameters, thereby accelerate the PINN-based development for molecular applications.