 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCoarse-Grained Boltzmann Generators
Feb 11, 2026Sampling equilibrium molecular configurations from the Boltzmann distribution is a longstanding challenge. Boltzmann Generators (BGs) address this by combining exact-likelihood generative models with importance sampling, but their practical scalability is limited. Meanwhile, coarse-grained surrogates enable the modeling of larger systems by reducing effective dimensionality, yet often lack the reweighting process required to ensure asymptotically correct statistics. In this work, we propose Coarse-Grained Boltzmann Generators (CG-BGs), a principled framework that unifies scalable reduced-order modeling with the exactness of importance sampling. CG-BGs act in a coarse-grained coordinate space, using a learned potential of mean force (PMF) to reweight samples generated by a flow-based model. Crucially, we show that this PMF can be efficiently learned from rapidly converged data via force matching. Our results demonstrate that CG-BGs faithfully capture complex interactions mediated by explicit solvent within highly reduced representations, establishing a scalable pathway for the unbiased sampling of larger molecular systems.
Generalization of Long-Range Machine Learning Potentials in Complex Chemical Spaces
Dec 08, 2025The vastness of chemical space makes generalization a central challenge in the development of machine learning interatomic potentials (MLIPs). While MLIPs could enable large-scale atomistic simulations with near-quantum accuracy, their usefulness is often limited by poor transferability to out-of-distribution samples. Here, we systematically evaluate different MLIP architectures with long-range corrections across diverse chemical spaces and show that such schemes are essential, not only for improving in-distribution performance but, more importantly, for enabling significant gains in transferability to unseen regions of chemical space. To enable a more rigorous benchmarking, we introduce biased train-test splitting strategies, which explicitly test the model performance in significantly different regions of chemical space. Together, our findings highlight the importance of long-range modeling for achieving generalizable MLIPs and provide a framework for diagnosing systematic failures across chemical space. Although we demonstrate our methodology on metal-organic frameworks, it is broadly applicable to other materials, offering insights into the design of more robust and transferable MLIPs.
chemtrain-deploy: A parallel and scalable framework for machine learning potentials in million-atom MD simulations
Jun 04, 2025Machine learning potentials (MLPs) have advanced rapidly and show great promise to transform molecular dynamics (MD) simulations. However, most existing software tools are tied to specific MLP architectures, lack integration with standard MD packages, or are not parallelizable across GPUs. To address these challenges, we present chemtrain-deploy, a framework that enables model-agnostic deployment of MLPs in LAMMPS. chemtrain-deploy supports any JAX-defined semi-local potential, allowing users to exploit the functionality of LAMMPS and perform large-scale MLP-based MD simulations on multiple GPUs. It achieves state-of-the-art efficiency and scales to systems containing millions of atoms. We validate its performance and scalability using graph neural network architectures, including MACE, Allegro, and PaiNN, applied to a variety of systems, such as liquid-vapor interfaces, crystalline materials, and solvated peptides. Our results highlight the practical utility of chemtrain-deploy for real-world, high-performance simulations and provide guidance for MLP architecture selection and future design.
JaxSGMC: Modular stochastic gradient MCMC in JAX
May 16, 2025We present JaxSGMC, an application-agnostic library for stochastic gradient Markov chain Monte Carlo (SG-MCMC) in JAX. SG-MCMC schemes are uncertainty quantification (UQ) methods that scale to large datasets and high-dimensional models, enabling trustworthy neural network predictions via Bayesian deep learning. JaxSGMC implements several state-of-the-art SG-MCMC samplers to promote UQ in deep learning by reducing the barriers of entry for switching from stochastic optimization to SG-MCMC sampling. Additionally, JaxSGMC allows users to build custom samplers from standard SG-MCMC building blocks. Due to this modular structure, we anticipate that JaxSGMC will accelerate research into novel SG-MCMC schemes and facilitate their application across a broad range of domains.
Enhancing Machine Learning Potentials through Transfer Learning across Chemical Elements
Feb 19, 2025Machine Learning Potentials (MLPs) can enable simulations of ab initio accuracy at orders of magnitude lower computational cost. However, their effectiveness hinges on the availability of considerable datasets to ensure robust generalization across chemical space and thermodynamic conditions. The generation of such datasets can be labor-intensive, highlighting the need for innovative methods to train MLPs in data-scarce scenarios. Here, we introduce transfer learning of potential energy surfaces between chemically similar elements. Specifically, we leverage the trained MLP for silicon to initialize and expedite the training of an MLP for germanium. Utilizing classical force field and ab initio datasets, we demonstrate that transfer learning surpasses traditional training from scratch in force prediction, leading to more stable simulations and improved temperature transferability. These advantages become even more pronounced as the training dataset size decreases. The out-of-target property analysis shows that transfer learning leads to beneficial but sometimes adversarial effects. Our findings demonstrate that transfer learning across chemical elements is a promising technique for developing accurate and numerically stable MLPs, particularly in a data-scarce regime.
Learning Non-Local Molecular Interactions via Equivariant Local Representations and Charge Equilibration
Jan 31, 2025



Graph Neural Network (GNN) potentials relying on chemical locality offer near-quantum mechanical accuracy at significantly reduced computational costs. By propagating local information to distance particles, Message-passing neural networks (MPNNs) extend the locality concept to model interactions beyond their local neighborhood. Still, this locality precludes modeling long-range effects, such as charge transfer, electrostatic interactions, and dispersion effects, which are critical to adequately describe many real-world systems. In this work, we propose the Charge Equilibration Layer for Long-range Interactions (CELLI) to address the challenging modeling of non-local interactions and the high computational cost of MPNNs. This novel architecture generalizes the fourth-generation high-dimensional neural network (4GHDNN) concept, integrating the charge equilibration (Qeq) method into a model-agnostic building block for modern equivariant GNN potentials. A series of benchmarks show that CELLI can extend the strictly local Allegro architecture to model highly non-local interactions and charge transfer. Our architecture generalizes to diverse datasets and large structures, achieving an accuracy comparable to MPNNs at about twice the computational efficiency.
chemtrain: Learning Deep Potential Models via Automatic Differentiation and Statistical Physics
Aug 28, 2024



Neural Networks (NNs) are promising models for refining the accuracy of molecular dynamics, potentially opening up new fields of application. Typically trained bottom-up, atomistic NN potential models can reach first-principle accuracy, while coarse-grained implicit solvent NN potentials surpass classical continuum solvent models. However, overcoming the limitations of costly generation of accurate reference data and data inefficiency of common bottom-up training demands efficient incorporation of data from many sources. This paper introduces the framework chemtrain to learn sophisticated NN potential models through customizable training routines and advanced training algorithms. These routines can combine multiple top-down and bottom-up algorithms, e.g., to incorporate both experimental and simulation data or pre-train potentials with less costly algorithms. chemtrain provides an object-oriented high-level interface to simplify the creation of custom routines. On the lower level, chemtrain relies on JAX to compute gradients and scale the computations to use available resources. We demonstrate the simplicity and importance of combining multiple algorithms in the examples of parametrizing an all-atomistic model of titanium and a coarse-grained implicit solvent model of alanine dipeptide.
Predicting solvation free energies with an implicit solvent machine learning potential
May 31, 2024



Machine learning (ML) potentials are a powerful tool in molecular modeling, enabling ab initio accuracy for comparably small computational costs. Nevertheless, all-atom simulations employing best-performing graph neural network architectures are still too expensive for applications requiring extensive sampling, such as free energy computations. Implicit solvent models could provide the necessary speed-up due to reduced degrees of freedom and faster dynamics. Here, we introduce a Solvation Free Energy Path Reweighting (ReSolv) framework to parametrize an implicit solvent ML potential for small organic molecules that accurately predicts the hydration free energy, an essential parameter in drug design and pollutant modeling. With a combination of top-down (experimental hydration free energy data) and bottom-up (ab initio data of molecules in a vacuum) learning, ReSolv bypasses the need for intractable ab initio data of molecules in explicit bulk solvent and does not have to resort to less accurate data-generating models. On the FreeSolv dataset, ReSolv achieves a mean absolute error close to average experimental uncertainty, significantly outperforming standard explicit solvent force fields. The presented framework paves the way toward deep molecular models that are more accurate yet computationally cheaper than classical atomistic models.
Accurate machine learning force fields via experimental and simulation data fusion
Aug 17, 2023Machine Learning (ML)-based force fields are attracting ever-increasing interest due to their capacity to span spatiotemporal scales of classical interatomic potentials at quantum-level accuracy. They can be trained based on high-fidelity simulations or experiments, the former being the common case. However, both approaches are impaired by scarce and erroneous data resulting in models that either do not agree with well-known experimental observations or are under-constrained and only reproduce some properties. Here we leverage both Density Functional Theory (DFT) calculations and experimentally measured mechanical properties and lattice parameters to train an ML potential of titanium. We demonstrate that the fused data learning strategy can concurrently satisfy all target objectives, thus resulting in a molecular model of higher accuracy compared to the models trained with a single data source. The inaccuracies of DFT functionals at target experimental properties were corrected, while the investigated off-target properties remained largely unperturbed. Our approach is applicable to any material and can serve as a general strategy to obtain highly accurate ML potentials.
Scalable Bayesian Uncertainty Quantification for Neural Network Potentials: Promise and Pitfalls
Dec 15, 2022
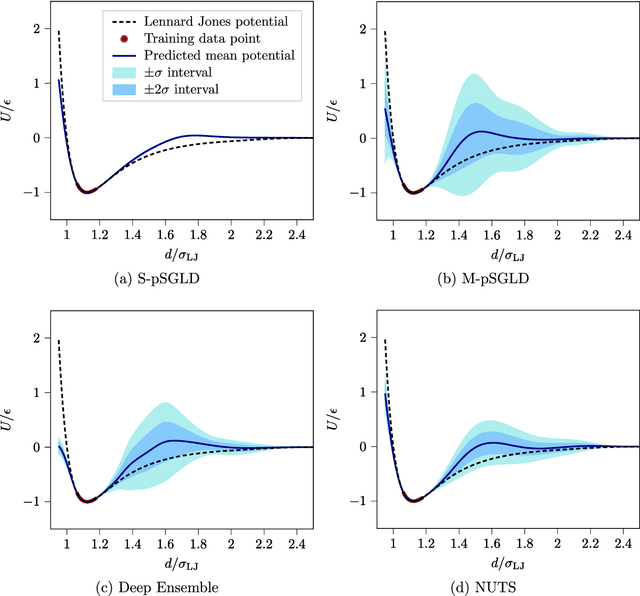
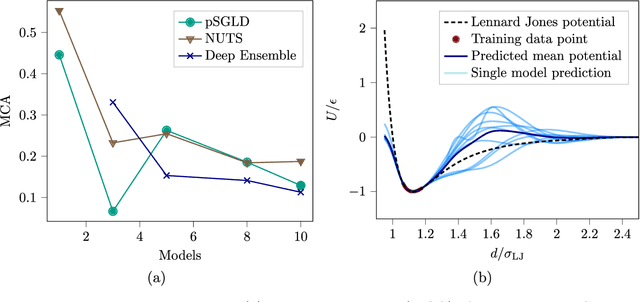
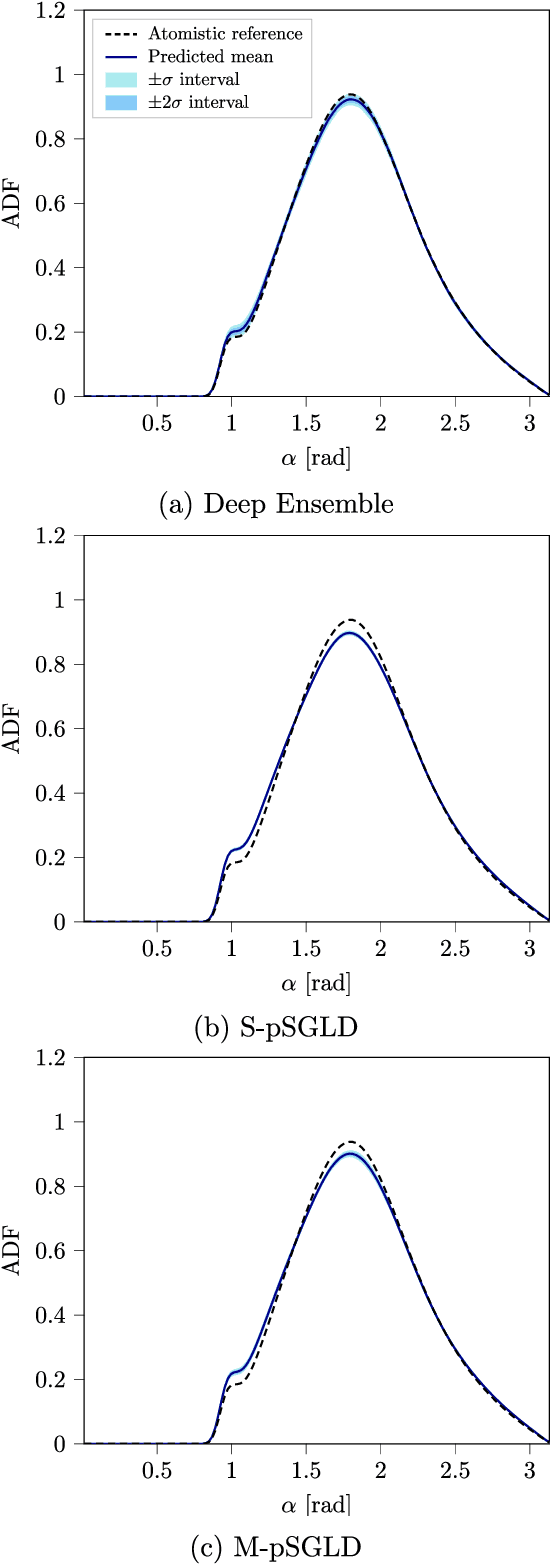
Neural network (NN) potentials promise highly accurate molecular dynamics (MD) simulations within the computational complexity of classical MD force fields. However, when applied outside their training domain, NN potential predictions can be inaccurate, increasing the need for Uncertainty Quantification (UQ). Bayesian modeling provides the mathematical framework for UQ, but classical Bayesian methods based on Markov chain Monte Carlo (MCMC) are computationally intractable for NN potentials. By training graph NN potentials for coarse-grained systems of liquid water and alanine dipeptide, we demonstrate here that scalable Bayesian UQ via stochastic gradient MCMC (SG-MCMC) yields reliable uncertainty estimates for MD observables. We show that cold posteriors can reduce the required training data size and that for reliable UQ, multiple Markov chains are needed. Additionally, we find that SG-MCMC and the Deep Ensemble method achieve comparable results, despite shorter training and less hyperparameter tuning of the latter. We show that both methods can capture aleatoric and epistemic uncertainty reliably, but not systematic uncertainty, which needs to be minimized by adequate modeling to obtain accurate credible intervals for MD observables. Our results represent a step towards accurate UQ that is of vital importance for trustworthy NN potential-based MD simulations required for decision-making in practice.