 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerated Simulations of Molecular Systems through Learning of their Effective Dynamics
Paper and Code
Feb 17, 2021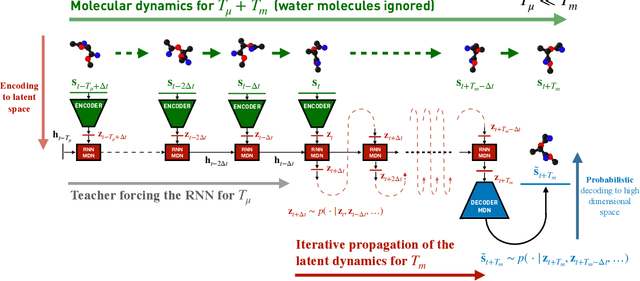
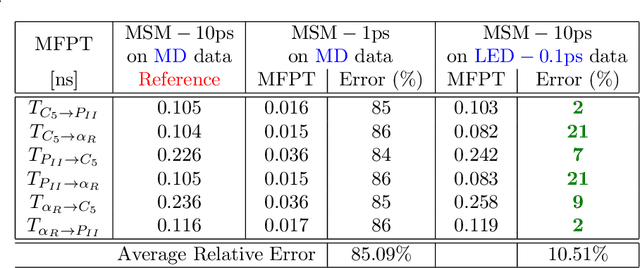
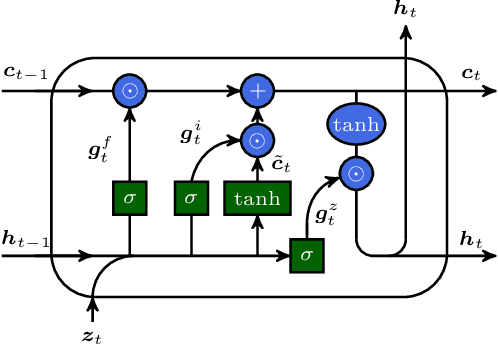
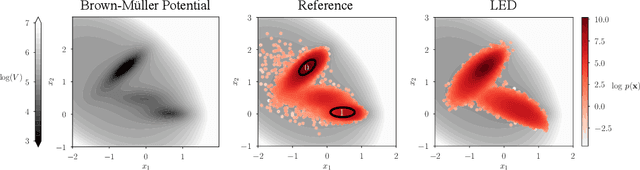
Simulations are vital for understanding and predicting the evolution of complex molecular systems. However, despite advances in algorithms and special purpose hardware, accessing the timescales necessary to capture the structural evolution of bio-molecules remains a daunting task. In this work we present a novel framework to advance simulation timescales by up to three orders of magnitude, by learning the effective dynamics (LED) of molecular systems. LED augments the equation-free methodology by employing a probabilistic mapping between coarse and fine scales using mixture density network (MDN) autoencoders and evolves the non-Markovian latent dynamics using long short-term memory MDNs. We demonstrate the effectiveness of LED in the M\"ueller-Brown potential, the Trp Cage protein, and the alanine dipeptide. LED identifies explainable reduced-order representations and can generate, at any instant, the respective all-atom molecular trajectories. We believe that the proposed framework provides a dramatic increase to simulation capabilities and opens new horizons for the effective modeling of complex molecular systems.