 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNot all tickets are equal and we know it: Guiding pruning with domain-specific knowledge
Mar 05, 2024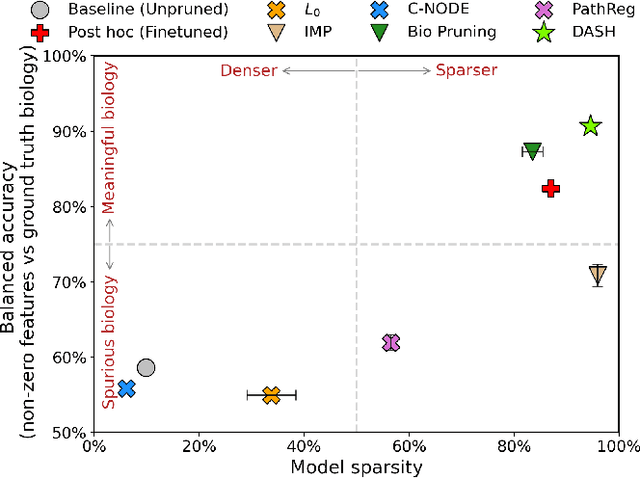

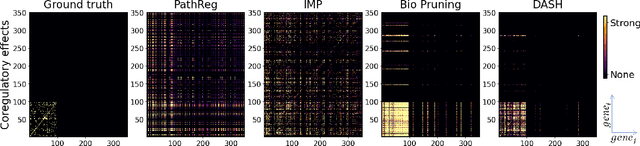

Neural structure learning is of paramount importance for scientific discovery and interpretability. Yet, contemporary pruning algorithms that focus on computational resource efficiency face algorithmic barriers to select a meaningful model that aligns with domain expertise. To mitigate this challenge, we propose DASH, which guides pruning by available domain-specific structural information. In the context of learning dynamic gene regulatory network models, we show that DASH combined with existing general knowledge on interaction partners provides data-specific insights aligned with biology. For this task, we show on synthetic data with ground truth information and two real world applications the effectiveness of DASH, which outperforms competing methods by a large margin and provides more meaningful biological insights. Our work shows that domain specific structural information bears the potential to improve model-derived scientific insights.
Scaling up Continuous-Time Markov Chains Helps Resolve Underspecification
Jul 06, 2021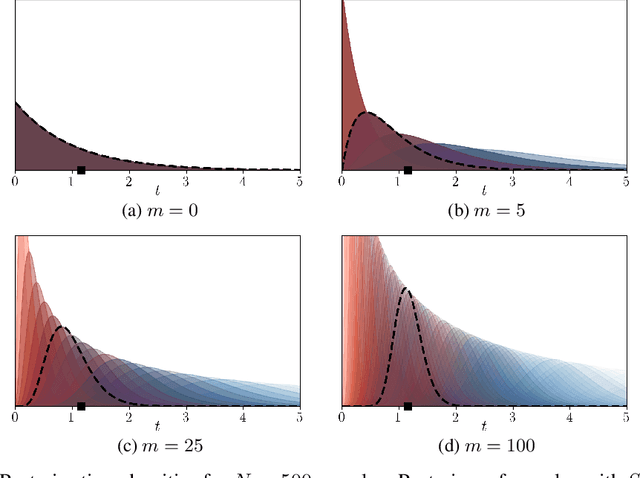

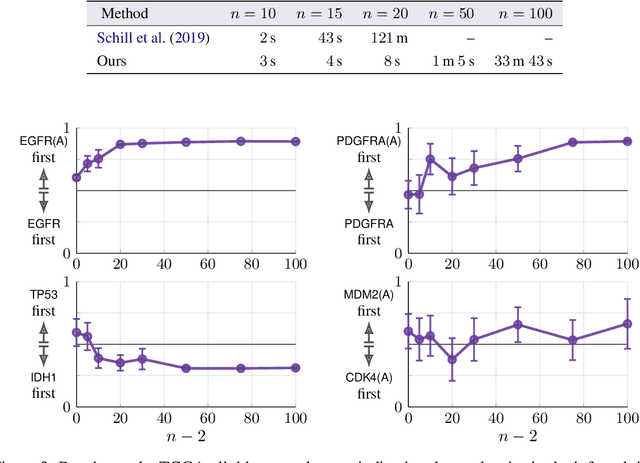
Modeling the time evolution of discrete sets of items (e.g., genetic mutations) is a fundamental problem in many biomedical applications. We approach this problem through the lens of continuous-time Markov chains, and show that the resulting learning task is generally underspecified in the usual setting of cross-sectional data. We explore a perhaps surprising remedy: including a number of additional independent items can help determine time order, and hence resolve underspecification. This is in sharp contrast to the common practice of limiting the analysis to a small subset of relevant items, which is followed largely due to poor scaling of existing methods. To put our theoretical insight into practice, we develop an approximate likelihood maximization method for learning continuous-time Markov chains, which can scale to hundreds of items and is orders of magnitude faster than previous methods. We demonstrate the effectiveness of our approach on synthetic and real cancer data.
Cascade Size Distributions and Why They Matter
Sep 09, 2019


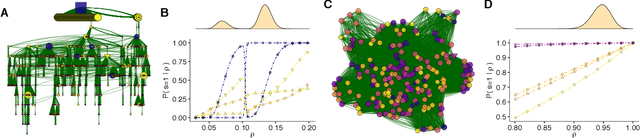
How likely is it that a few initial node activations are amplified to produce large response cascades that span a considerable part of an entire network? Our answer to this question relies on the Independent Cascade Model for weighted directed networks. In using this model, most of our insights have been derived from the study of average effects. Here, we shift the focus on the full probability distribution of the final cascade size. This shift allows us to explore both typical cascade outcomes and improbable but relevant extreme events. We present an efficient message passing algorithm to compute the final cascade size distribution and activation probabilities of nodes conditional on the final cascade size. Our approach is exact on trees but can be applied to any network topology. It approximates locally tree-like networks well and can lead to surprisingly good performance on more dense networks, as we show using real world data, including a miRNA- miRNA probabilistic interaction network for gastrointestinal cancer. We demonstrate the utility of our algorithms for clustering of nodes according to their functionality and influence maximization.