 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFew-Shot Graph Learning for Molecular Property Prediction
Feb 16, 2021
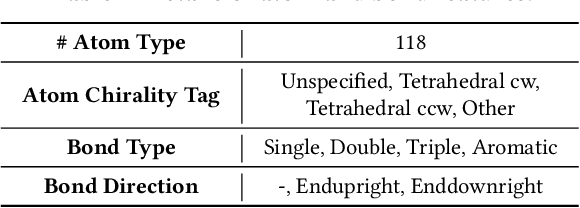


The recent success of graph neural networks has significantly boosted molecular property prediction, advancing activities such as drug discovery. The existing deep neural network methods usually require large training dataset for each property, impairing their performances in cases (especially for new molecular properties) with a limited amount of experimental data, which are common in real situations. To this end, we propose Meta-MGNN, a novel model for few-shot molecular property prediction. Meta-MGNN applies molecular graph neural network to learn molecular representation and builds a meta-learning framework for model optimization. To exploit unlabeled molecular information and address task heterogeneity of different molecular properties, Meta-MGNN further incorporates molecular structure, attribute based self-supervised modules and self-attentive task weights into the former framework, strengthening the whole learning model. Extensive experiments on two public multi-property datasets demonstrate that Meta-MGNN outperforms a variety of state-of-the-art methods.