 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA multimodal ensemble approach for clear cell renal cell carcinoma treatment outcome prediction
Dec 10, 2024

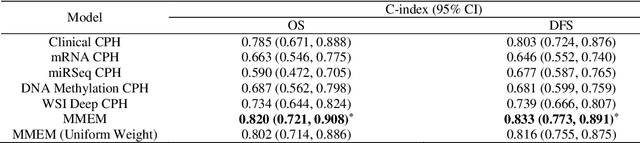
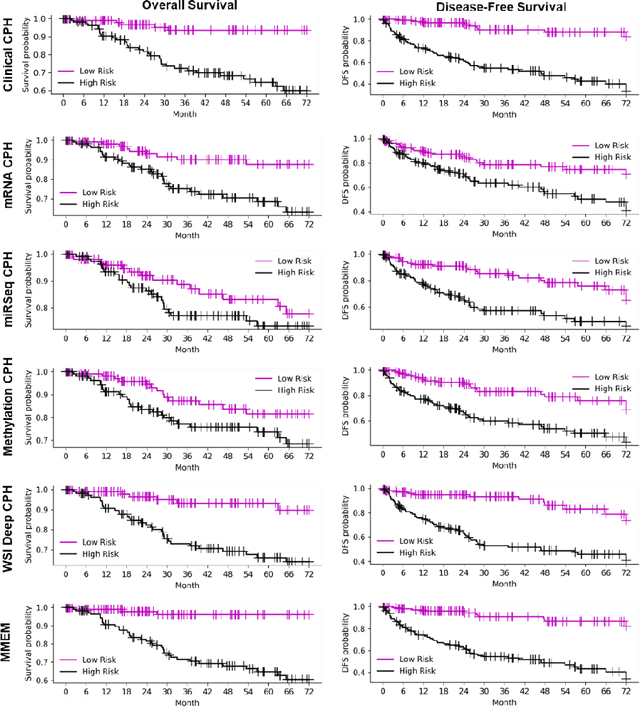
Purpose: A reliable cancer prognosis model for clear cell renal cell carcinoma (ccRCC) can enhance personalized treatment. We developed a multi-modal ensemble model (MMEM) that integrates pretreatment clinical data, multi-omics data, and histopathology whole slide image (WSI) data to predict overall survival (OS) and disease-free survival (DFS) for ccRCC patients. Methods: We analyzed 226 patients from The Cancer Genome Atlas Kidney Renal Clear Cell Carcinoma (TCGA-KIRC) dataset, which includes OS, DFS follow-up data, and five data modalities: clinical data, WSIs, and three multi-omics datasets (mRNA, miRNA, and DNA methylation). Separate survival models were built for OS and DFS. Cox-proportional hazards (CPH) model with forward feature selection is used for clinical and multi-omics data. Features from WSIs were extracted using ResNet and three general-purpose foundation models. A deep learning-based CPH model predicted survival using encoded WSI features. Risk scores from all models were combined based on training performance. Results: Performance was assessed using concordance index (C-index) and AUROC. The clinical feature-based CPH model received the highest weight for both OS and DFS tasks. Among WSI-based models, the general-purpose foundation model (UNI) achieved the best performance. The final MMEM model surpassed single-modality models, achieving C-indices of 0.820 (OS) and 0.833 (DFS), and AUROC values of 0.831 (3-year patient death) and 0.862 (cancer recurrence). Using predicted risk medians to stratify high- and low-risk groups, log-rank tests showed improved performance in both OS and DFS compared to single-modality models. Conclusion: MMEM is the first multi-modal model for ccRCC patients, integrating five data modalities. It outperformed single-modality models in prognostic ability and has the potential to assist in ccRCC patient management if independently validated.
Histopathology Based AI Model Predicts Anti-Angiogenic Therapy Response in Renal Cancer Clinical Trial
May 28, 2024Predictive biomarkers of treatment response are lacking for metastatic clear cell renal cell carcinoma (ccRCC), a tumor type that is treated with angiogenesis inhibitors, immune checkpoint inhibitors, mTOR inhibitors and a HIF2 inhibitor. The Angioscore, an RNA-based quantification of angiogenesis, is arguably the best candidate to predict anti-angiogenic (AA) response. However, the clinical adoption of transcriptomic assays faces several challenges including standardization, time delay, and high cost. Further, ccRCC tumors are highly heterogenous, and sampling multiple areas for sequencing is impractical. Here we present a novel deep learning (DL) approach to predict the Angioscore from ubiquitous histopathology slides. To overcome the lack of interpretability, one of the biggest limitations of typical DL models, our model produces a visual vascular network which is the basis of the model's prediction. To test its reliability, we applied this model to multiple cohorts including a clinical trial dataset. Our model accurately predicts the RNA-based Angioscore on multiple independent cohorts (spearman correlations of 0.77 and 0.73). Further, the predictions help unravel meaningful biology such as association of angiogenesis with grade, stage, and driver mutation status. Finally, we find our model can predict response to AA therapy, in both a real-world cohort and the IMmotion150 clinical trial. The predictive power of our model vastly exceeds that of CD31, a marker of vasculature, and nearly rivals the performance (c-index 0.66 vs 0.67) of the ground truth RNA-based Angioscore at a fraction of the cost. By providing a robust yet interpretable prediction of the Angioscore from histopathology slides alone, our approach offers insights into angiogenesis biology and AA treatment response.