 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLightweight Optimal-Transport Harmonization on Edge Devices
Nov 16, 2025Color harmonization adjusts the colors of an inserted object so that it perceptually matches the surrounding image, resulting in a seamless composite. The harmonization problem naturally arises in augmented reality (AR), yet harmonization algorithms are not currently integrated into AR pipelines because real-time solutions are scarce. In this work, we address color harmonization for AR by proposing a lightweight approach that supports on-device inference. For this, we leverage classical optimal transport theory by training a compact encoder to predict the Monge-Kantorovich transport map. We benchmark our MKL-Harmonizer algorithm against state-of-the-art methods and demonstrate that for real composite AR images our method achieves the best aggregated score. We release our dedicated AR dataset of composite images with pixel-accurate masks and data-gathering toolkit to support further data acquisition by researchers.
Dock2D: Synthetic data for the molecular recognition problem
Dec 07, 2022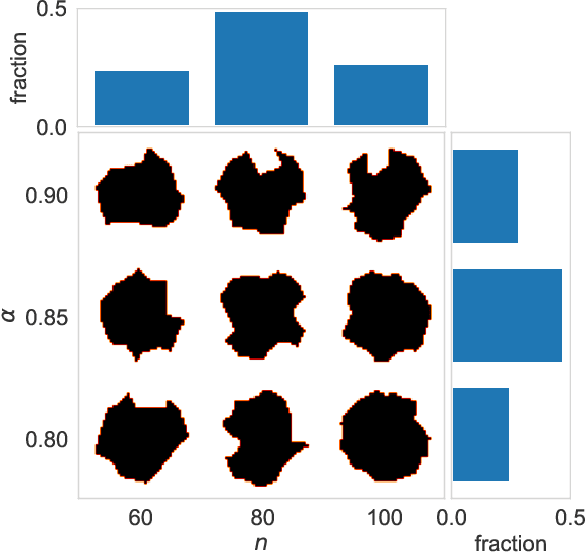



Predicting the physical interaction of proteins is a cornerstone problem in computational biology. New classes of learning-based algorithms are actively being developed, and are typically trained end-to-end on protein complex structures extracted from the Protein Data Bank. These training datasets tend to be large and difficult to use for prototyping and, unlike image or natural language datasets, they are not easily interpretable by non-experts. We present Dock2D-IP and Dock2D-IF, two "toy" datasets that can be used to select algorithms predicting protein-protein interactions$\unicode{x2014}$or any other type of molecular interactions. Using two-dimensional shapes as input, each example from Dock2D-IP ("interaction pose") describes the interaction pose of two shapes known to interact and each example from Dock2D-IF ("interaction fact") describes whether two shapes form a stable complex or not. We propose a number of baseline solutions to the problem and show that the same underlying energy function can be learned either by solving the interaction pose task (formulated as an energy-minimization "docking" problem) or the fact-of-interaction task (formulated as a binding free energy estimation problem).
TorchProteinLibrary: A computationally efficient, differentiable representation of protein structure
Nov 23, 2018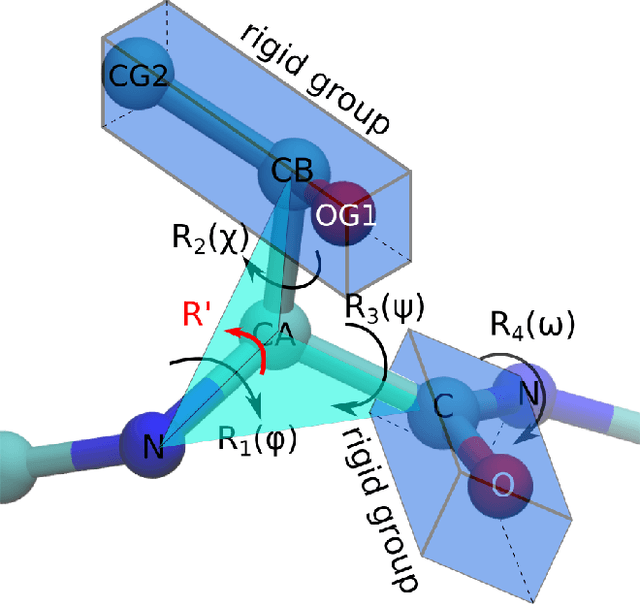
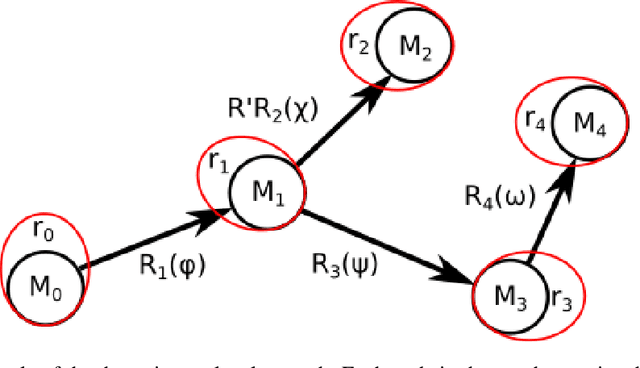
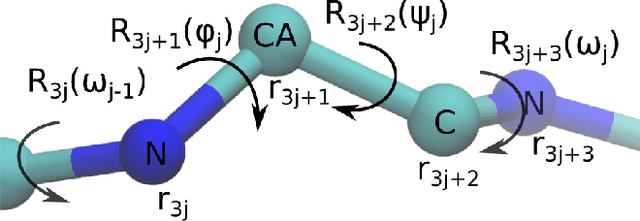
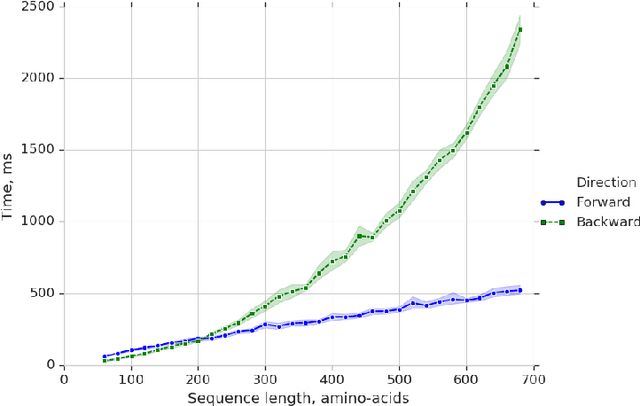
Predicting the structure of a protein from its sequence is a cornerstone task of molecular biology. Established methods in the field, such as homology modeling and fragment assembly, appeared to have reached their limit. However, this year saw the emergence of promising new approaches: end-to-end protein structure and dynamics models, as well as reinforcement learning applied to protein folding. For these approaches to be investigated on a larger scale, an efficient implementation of their key computational primitives is required. In this paper we present a library of differentiable mappings from two standard dihedral-angle representations of protein structure (full-atom representation "$\phi,\psi,\omega,\chi$" and backbone-only representation "$\phi,\psi,\omega$") to atomic Cartesian coordinates. The source code and documentation can be found at https://github.com/lupoglaz/TorchProteinLibrary.
Towards Gene Expression Convolutions using Gene Interaction Graphs
Jun 18, 2018
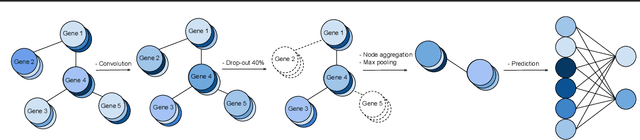
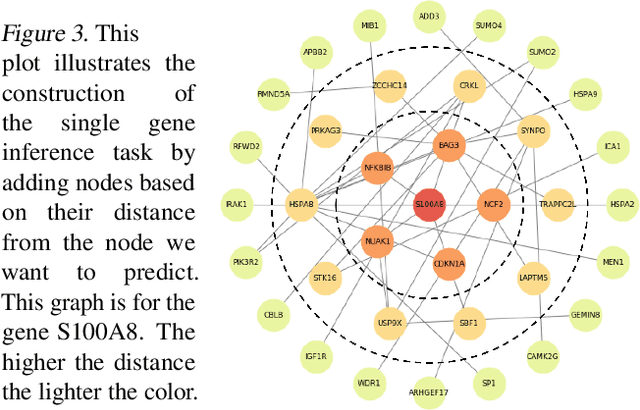
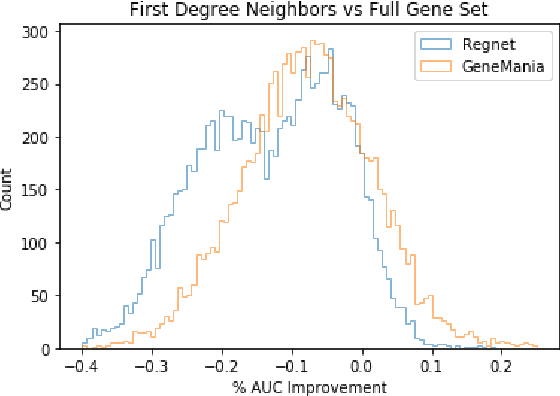
We study the challenges of applying deep learning to gene expression data. We find experimentally that there exists non-linear signal in the data, however is it not discovered automatically given the noise and low numbers of samples used in most research. We discuss how gene interaction graphs (same pathway, protein-protein, co-expression, or research paper text association) can be used to impose a bias on a deep model similar to the spatial bias imposed by convolutions on an image. We explore the usage of Graph Convolutional Neural Networks coupled with dropout and gene embeddings to utilize the graph information. We find this approach provides an advantage for particular tasks in a low data regime but is very dependent on the quality of the graph used. We conclude that more work should be done in this direction. We design experiments that show why existing methods fail to capture signal that is present in the data when features are added which clearly isolates the problem that needs to be addressed.