 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards Gene Expression Convolutions using Gene Interaction Graphs
Paper and Code
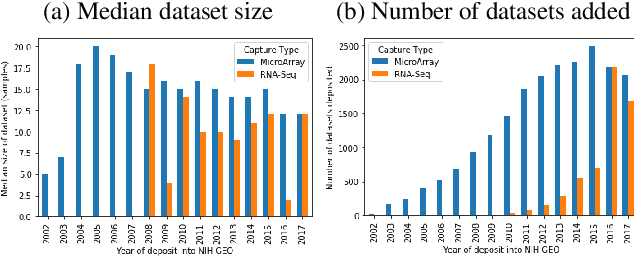
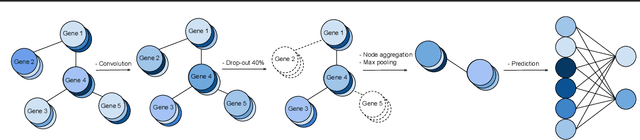
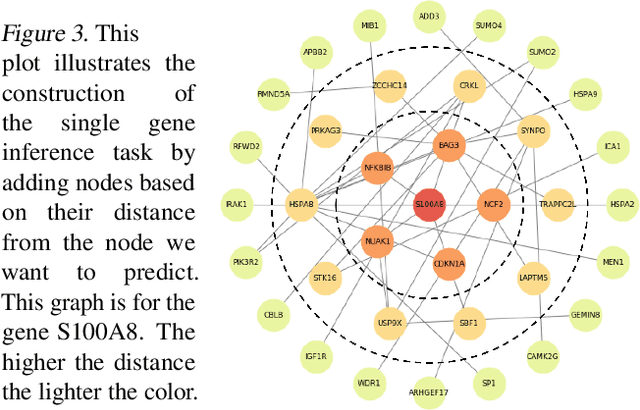
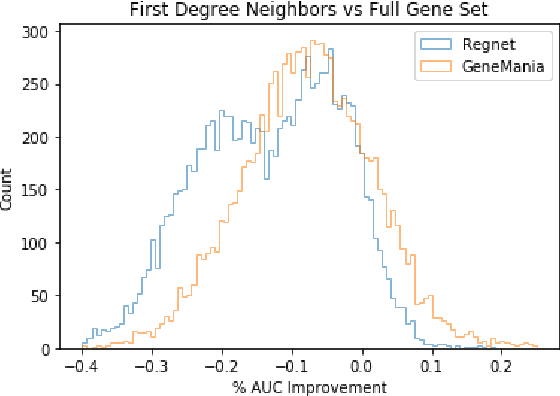
We study the challenges of applying deep learning to gene expression data. We find experimentally that there exists non-linear signal in the data, however is it not discovered automatically given the noise and low numbers of samples used in most research. We discuss how gene interaction graphs (same pathway, protein-protein, co-expression, or research paper text association) can be used to impose a bias on a deep model similar to the spatial bias imposed by convolutions on an image. We explore the usage of Graph Convolutional Neural Networks coupled with dropout and gene embeddings to utilize the graph information. We find this approach provides an advantage for particular tasks in a low data regime but is very dependent on the quality of the graph used. We conclude that more work should be done in this direction. We design experiments that show why existing methods fail to capture signal that is present in the data when features are added which clearly isolates the problem that needs to be addressed.