 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMoRE-GNN: Multi-omics Data Integration with a Heterogeneous Graph Autoencoder
Oct 08, 2025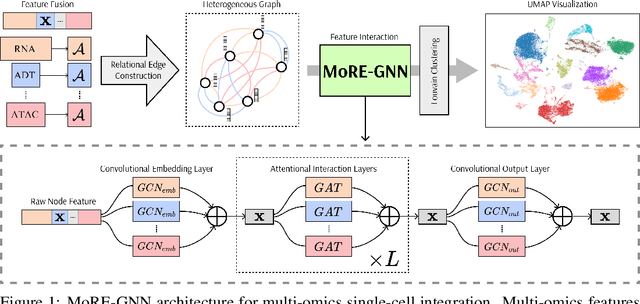

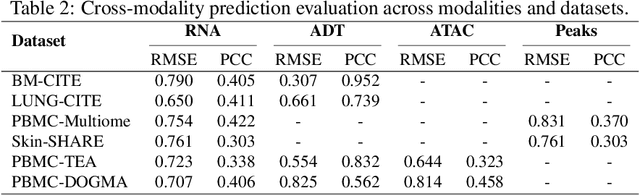
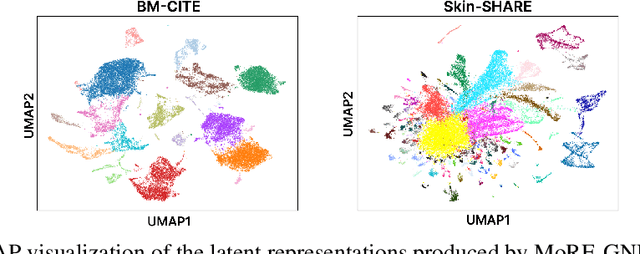
The integration of multi-omics single-cell data remains challenging due to high-dimensionality and complex inter-modality relationships. To address this, we introduce MoRE-GNN (Multi-omics Relational Edge Graph Neural Network), a heterogeneous graph autoencoder that combines graph convolution and attention mechanisms to dynamically construct relational graphs directly from data. Evaluations on six publicly available datasets demonstrate that MoRE-GNN captures biologically meaningful relationships and outperforms existing methods, particularly in settings with strong inter-modality correlations. Furthermore, the learned representations allow for accurate downstream cross-modal predictions. While performance may vary with dataset complexity, MoRE-GNN offers an adaptive, scalable and interpretable framework for advancing multi-omics integration.
Neural Embeddings for Protein Graphs
Jun 07, 2023



Proteins perform much of the work in living organisms, and consequently the development of efficient computational methods for protein representation is essential for advancing large-scale biological research. Most current approaches struggle to efficiently integrate the wealth of information contained in the protein sequence and structure. In this paper, we propose a novel framework for embedding protein graphs in geometric vector spaces, by learning an encoder function that preserves the structural distance between protein graphs. Utilizing Graph Neural Networks (GNNs) and Large Language Models (LLMs), the proposed framework generates structure- and sequence-aware protein representations. We demonstrate that our embeddings are successful in the task of comparing protein structures, while providing a significant speed-up compared to traditional approaches based on structural alignment. Our framework achieves remarkable results in the task of protein structure classification; in particular, when compared to other work, the proposed method shows an average F1-Score improvement of 26% on out-of-distribution (OOD) samples and of 32% when tested on samples coming from the same distribution as the training data. Our approach finds applications in areas such as drug prioritization, drug re-purposing, disease sub-type analysis and elsewhere.
CIN++: Enhancing Topological Message Passing
Jun 06, 2023Graph Neural Networks (GNNs) have demonstrated remarkable success in learning from graph-structured data. However, they face significant limitations in expressive power, struggling with long-range interactions and lacking a principled approach to modeling higher-order structures and group interactions. Cellular Isomorphism Networks (CINs) recently addressed most of these challenges with a message passing scheme based on cell complexes. Despite their advantages, CINs make use only of boundary and upper messages which do not consider a direct interaction between the rings present in the underlying complex. Accounting for these interactions might be crucial for learning representations of many real-world complex phenomena such as the dynamics of supramolecular assemblies, neural activity within the brain, and gene regulation processes. In this work, we propose CIN++, an enhancement of the topological message passing scheme introduced in CINs. Our message passing scheme accounts for the aforementioned limitations by letting the cells to receive also lower messages within each layer. By providing a more comprehensive representation of higher-order and long-range interactions, our enhanced topological message passing scheme achieves state-of-the-art results on large-scale and long-range chemistry benchmarks.
Deep reinforcement learning for quantum multiparameter estimation
Sep 01, 2022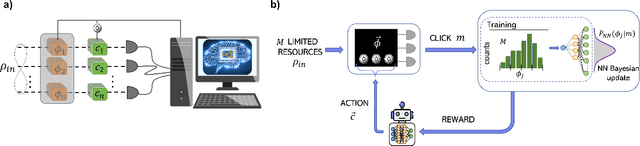
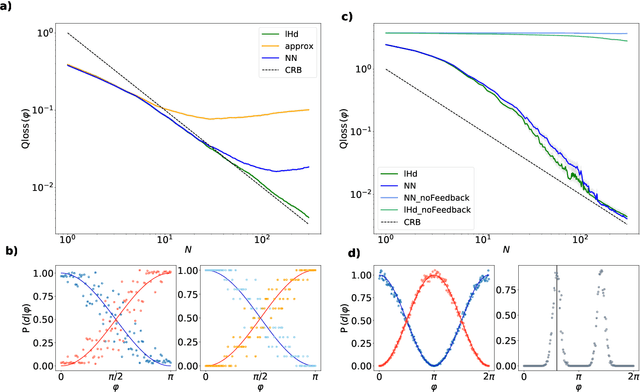
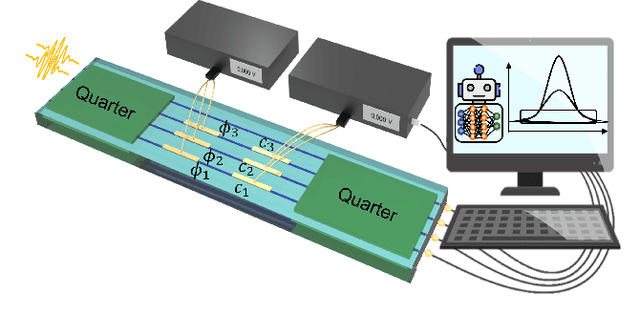
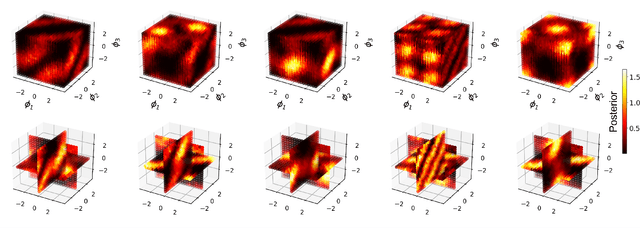
Estimation of physical quantities is at the core of most scientific research and the use of quantum devices promises to enhance its performances. In real scenarios, it is fundamental to consider that the resources are limited and Bayesian adaptive estimation represents a powerful approach to efficiently allocate, during the estimation process, all the available resources. However, this framework relies on the precise knowledge of the system model, retrieved with a fine calibration that often results computationally and experimentally demanding. Here, we introduce a model-free and deep learning-based approach to efficiently implement realistic Bayesian quantum metrology tasks accomplishing all the relevant challenges, without relying on any a-priori knowledge on the system. To overcome this need, a neural network is trained directly on experimental data to learn the multiparameter Bayesian update. Then, the system is set at its optimal working point through feedbacks provided by a reinforcement learning algorithm trained to reconstruct and enhance experiment heuristics of the investigated quantum sensor. Notably, we prove experimentally the achievement of higher estimation performances than standard methods, demonstrating the strength of the combination of these two black-box algorithms on an integrated photonic circuit. This work represents an important step towards fully artificial intelligence-based quantum metrology.
Rxn Hypergraph: a Hypergraph Attention Model for Chemical Reaction Representation
Jan 02, 2022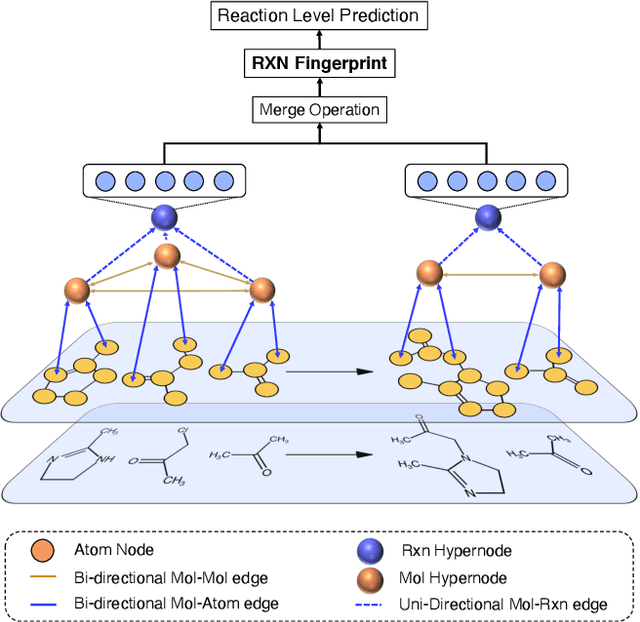
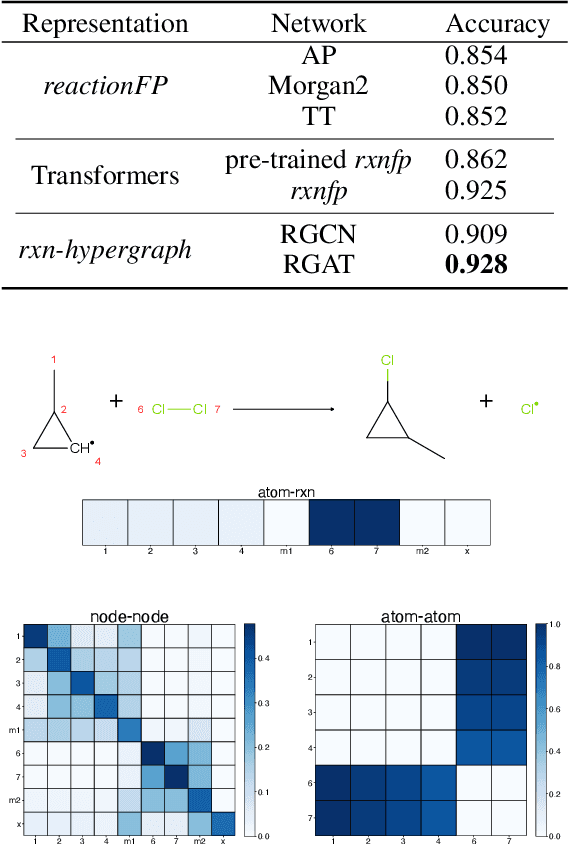
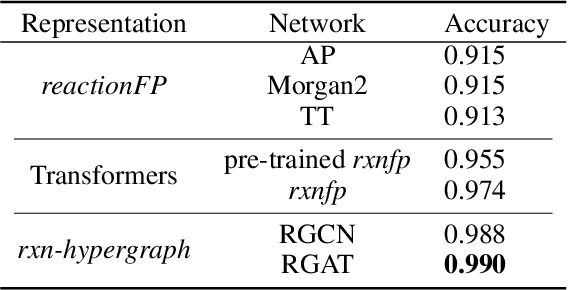
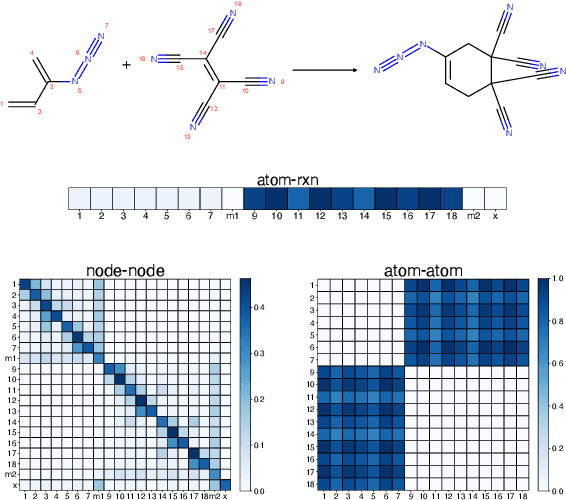
It is fundamental for science and technology to be able to predict chemical reactions and their properties. To achieve such skills, it is important to develop good representations of chemical reactions, or good deep learning architectures that can learn such representations automatically from the data. There is currently no universal and widely adopted method for robustly representing chemical reactions. Most existing methods suffer from one or more drawbacks, such as: (1) lacking universality; (2) lacking robustness; (3) lacking interpretability; or (4) requiring excessive manual pre-processing. Here we exploit graph-based representations of molecular structures to develop and test a hypergraph attention neural network approach to solve at once the reaction representation and property-prediction problems, alleviating the aforementioned drawbacks. We evaluate this hypergraph representation in three experiments using three independent data sets of chemical reactions. In all experiments, the hypergraph-based approach matches or outperforms other representations and their corresponding models of chemical reactions while yielding interpretable multi-level representations.