 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStructure-aware generation of drug-like molecules
Nov 07, 2021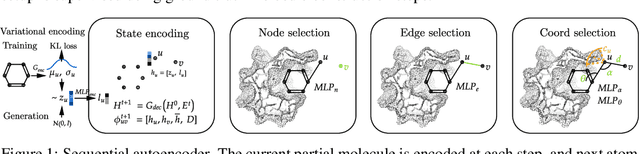
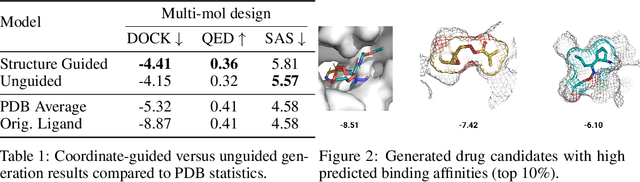
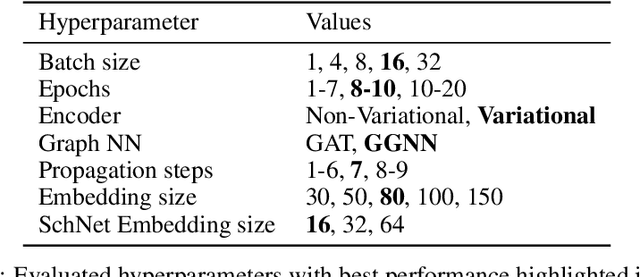
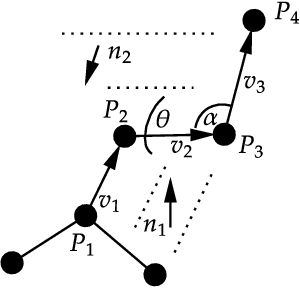
Structure-based drug design involves finding ligand molecules that exhibit structural and chemical complementarity to protein pockets. Deep generative methods have shown promise in proposing novel molecules from scratch (de-novo design), avoiding exhaustive virtual screening of chemical space. Most generative de-novo models fail to incorporate detailed ligand-protein interactions and 3D pocket structures. We propose a novel supervised model that generates molecular graphs jointly with 3D pose in a discretised molecular space. Molecules are built atom-by-atom inside pockets, guided by structural information from crystallographic data. We evaluate our model using a docking benchmark and find that guided generation improves predicted binding affinities by 8% and drug-likeness scores by 10% over the baseline. Furthermore, our model proposes molecules with binding scores exceeding some known ligands, which could be useful in future wet-lab studies.
On Graph Neural Network Ensembles for Large-Scale Molecular Property Prediction
Jun 29, 2021
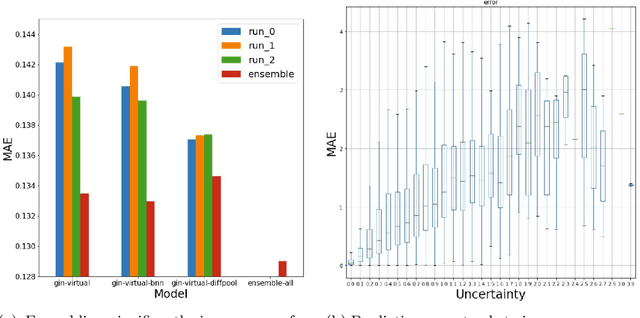
In order to advance large-scale graph machine learning, the Open Graph Benchmark Large Scale Challenge (OGB-LSC) was proposed at the KDD Cup 2021. The PCQM4M-LSC dataset defines a molecular HOMO-LUMO property prediction task on about 3.8M graphs. In this short paper, we show our current work-in-progress solution which builds an ensemble of three graph neural networks models based on GIN, Bayesian Neural Networks and DiffPool. Our approach outperforms the provided baseline by 7.6%. Moreover, using uncertainty in our ensemble's prediction, we can identify molecules whose HOMO-LUMO gaps are harder to predict (with Pearson's correlation of 0.5181). We anticipate that this will facilitate active learning.