 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConRetroBert: EMA Stabilized Dual Encoders for Template-Based Single-Step Retrosynthesis
May 12, 2026Template based single step retrosynthesis predicts reactants by selecting and applying an explicit reaction template, making each prediction traceable to a chemical transformation rule. This is useful for synthesis planning, but template based methods are often viewed as less competitive than template free models because template prediction is commonly formulated as global classification over a long tailed rule library. We argue that this weakness is not inherent to templates, but to the learning formulation. We present ConRetroBert, a dual encoder framework that reframes template based retrosynthesis as dense product template retrieval followed by candidate set listwise ranking. Stage 1 uses contrastive pretraining to learn a shared embedding space between products and reaction templates. Stage 2 refines template ranking over mined hard negative candidate sets with a multi positive listwise objective. To enable template side adaptation without destabilizing hard negative mining, ConRetroBert uses a slow moving exponential moving average template encoder for retrieval bank construction while updating the live template encoder through the ranking loss. On the local USPTO-50k benchmark, Stage 2 candidate set ranking improves top-1 reaction accuracy from 50.5% to 61.3%, while EMA stabilized template adaptation further improves it to 62.4%. Fine tuning from a leakage controlled USPTO-Full checkpoint reaches 75.4% top-1 accuracy on USPTO-50k. We also show that retrieval based template prediction is strong in the long tail of rare templates, and that many correct reactant predictions arise from alternative explicit templates rather than only the recorded positive label. Code and data are available at https://github.com/JahidBasher/ConRetroBert.
FairContrast: Enhancing Fairness through Contrastive learning and Customized Augmenting Methods on Tabular Data
Oct 02, 2025As AI systems become more embedded in everyday life, the development of fair and unbiased models becomes more critical. Considering the social impact of AI systems is not merely a technical challenge but a moral imperative. As evidenced in numerous research studies, learning fair and robust representations has proven to be a powerful approach to effectively debiasing algorithms and improving fairness while maintaining essential information for prediction tasks. Representation learning frameworks, particularly those that utilize self-supervised and contrastive learning, have demonstrated superior robustness and generalizability across various domains. Despite the growing interest in applying these approaches to tabular data, the issue of fairness in these learned representations remains underexplored. In this study, we introduce a contrastive learning framework specifically designed to address bias and learn fair representations in tabular datasets. By strategically selecting positive pair samples and employing supervised and self-supervised contrastive learning, we significantly reduce bias compared to existing state-of-the-art contrastive learning models for tabular data. Our results demonstrate the efficacy of our approach in mitigating bias with minimum trade-off in accuracy and leveraging the learned fair representations in various downstream tasks.
Equi-mRNA: Protein Translation Equivariant Encoding for mRNA Language Models
Aug 20, 2025The growing importance of mRNA therapeutics and synthetic biology highlights the need for models that capture the latent structure of synonymous codon (different triplets encoding the same amino acid) usage, which subtly modulates translation efficiency and gene expression. While recent efforts incorporate codon-level inductive biases through auxiliary objectives, they often fall short of explicitly modeling the structured relationships that arise from the genetic code's inherent symmetries. We introduce Equi-mRNA, the first codon-level equivariant mRNA language model that explicitly encodes synonymous codon symmetries as cyclic subgroups of 2D Special Orthogonal matrix (SO(2)). By combining group-theoretic priors with an auxiliary equivariance loss and symmetry-aware pooling, Equi-mRNA learns biologically grounded representations that outperform vanilla baselines across multiple axes. On downstream property-prediction tasks including expression, stability, and riboswitch switching Equi-mRNA delivers up to approximately 10% improvements in accuracy. In sequence generation, it produces mRNA constructs that are up to approximately 4x more realistic under Frechet BioDistance metrics and approximately 28% better preserve functional properties compared to vanilla baseline. Interpretability analyses further reveal that learned codon-rotation distributions recapitulate known GC-content biases and tRNA abundance patterns, offering novel insights into codon usage. Equi-mRNA establishes a new biologically principled paradigm for mRNA modeling, with significant implications for the design of next-generation therapeutics.
BoKDiff: Best-of-K Diffusion Alignment for Target-Specific 3D Molecule Generation
Jan 26, 2025Structure-based drug design (SBDD) leverages the 3D structure of biomolecular targets to guide the creation of new therapeutic agents. Recent advances in generative models, including diffusion models and geometric deep learning, have demonstrated promise in optimizing ligand generation. However, the scarcity of high-quality protein-ligand complex data and the inherent challenges in aligning generated ligands with target proteins limit the effectiveness of these methods. We propose BoKDiff, a novel framework that enhances ligand generation by combining multi-objective optimization and Best-of-K alignment methodologies. Built upon the DecompDiff model, BoKDiff generates diverse candidates and ranks them using a weighted evaluation of molecular properties such as QED, SA, and docking scores. To address alignment challenges, we introduce a method that relocates the center of mass of generated ligands to their docking poses, enabling accurate sub-component extraction. Additionally, we integrate a Best-of-N (BoN) sampling approach, which selects the optimal ligand from multiple generated candidates without requiring fine-tuning. BoN achieves exceptional results, with QED values exceeding 0.6, SA scores above 0.75, and a success rate surpassing 35%, demonstrating its efficiency and practicality. BoKDiff achieves state-of-the-art results on the CrossDocked2020 dataset, including a -8.58 average Vina docking score and a 26% success rate in molecule generation. This study is the first to apply Best-of-K alignment and Best-of-N sampling to SBDD, highlighting their potential to bridge generative modeling with practical drug discovery requirements. The code is provided at https://github.com/khodabandeh-ali/BoKDiff.git.
Fair Bilevel Neural Network (FairBiNN): On Balancing fairness and accuracy via Stackelberg Equilibrium
Oct 21, 2024



The persistent challenge of bias in machine learning models necessitates robust solutions to ensure parity and equal treatment across diverse groups, particularly in classification tasks. Current methods for mitigating bias often result in information loss and an inadequate balance between accuracy and fairness. To address this, we propose a novel methodology grounded in bilevel optimization principles. Our deep learning-based approach concurrently optimizes for both accuracy and fairness objectives, and under certain assumptions, achieving proven Pareto optimal solutions while mitigating bias in the trained model. Theoretical analysis indicates that the upper bound on the loss incurred by this method is less than or equal to the loss of the Lagrangian approach, which involves adding a regularization term to the loss function. We demonstrate the efficacy of our model primarily on tabular datasets such as UCI Adult and Heritage Health. When benchmarked against state-of-the-art fairness methods, our model exhibits superior performance, advancing fairness-aware machine learning solutions and bridging the accuracy-fairness gap. The implementation of FairBiNN is available on https://github.com/yazdanimehdi/FairBiNN.
FragXsiteDTI: Revealing Responsible Segments in Drug-Target Interaction with Transformer-Driven Interpretation
Nov 04, 2023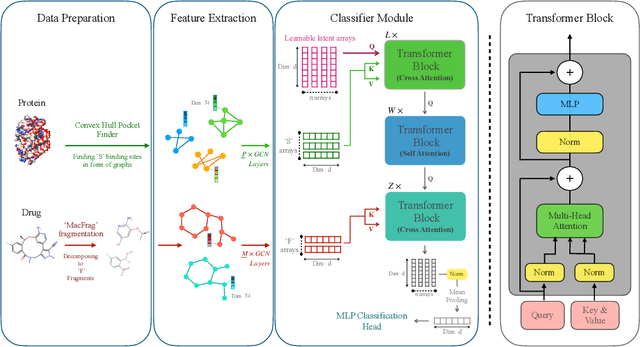
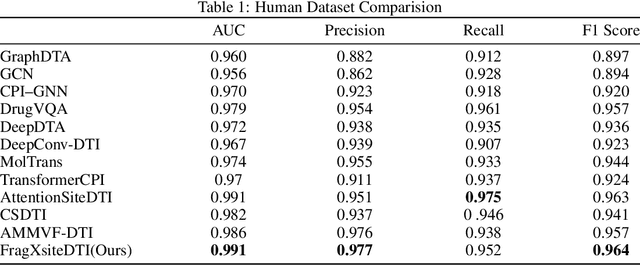
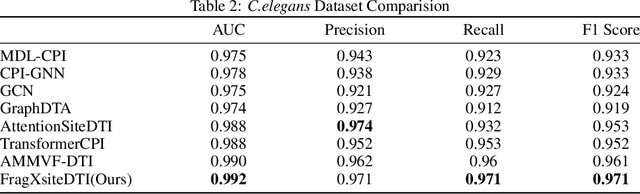
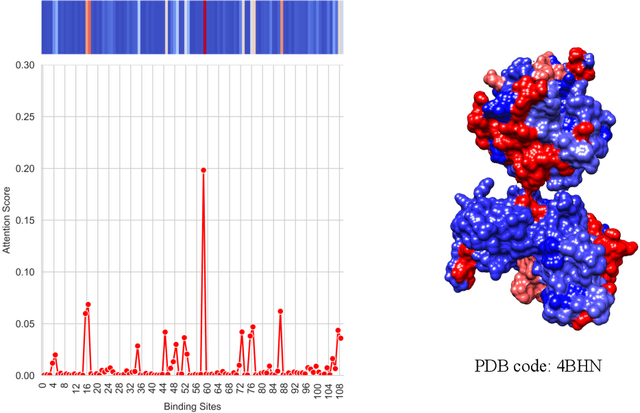
Drug-Target Interaction (DTI) prediction is vital for drug discovery, yet challenges persist in achieving model interpretability and optimizing performance. We propose a novel transformer-based model, FragXsiteDTI, that aims to address these challenges in DTI prediction. Notably, FragXsiteDTI is the first DTI model to simultaneously leverage drug molecule fragments and protein pockets. Our information-rich representations for both proteins and drugs offer a detailed perspective on their interaction. Inspired by the Perceiver IO framework, our model features a learnable latent array, initially interacting with protein binding site embeddings using cross-attention and later refined through self-attention and used as a query to the drug fragments in the drug's cross-attention transformer block. This learnable query array serves as a mediator and enables seamless information translation, preserving critical nuances in drug-protein interactions. Our computational results on three benchmarking datasets demonstrate the superior predictive power of our model over several state-of-the-art models. We also show the interpretability of our model in terms of the critical components of both target proteins and drug molecules within drug-target pairs.