 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBenchmarking Machine Learning Robustness in Covid-19 Genome Sequence Classification
Jul 18, 2022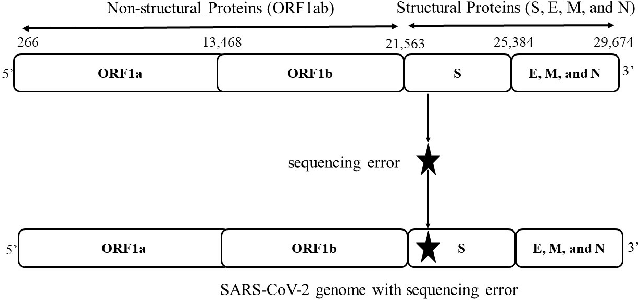
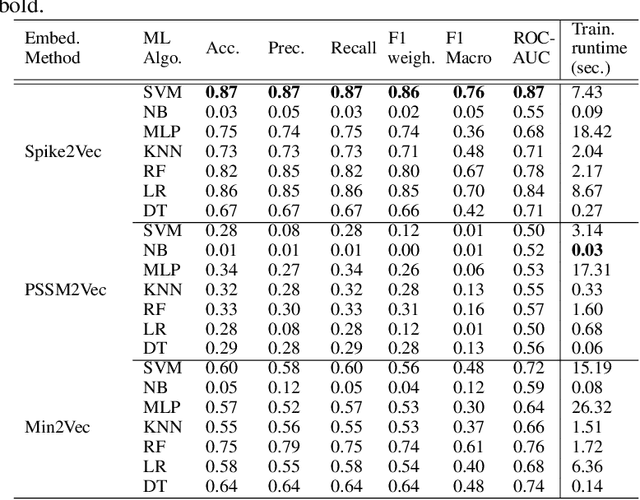
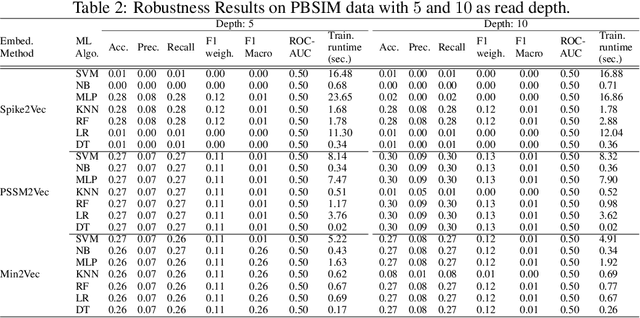
The rapid spread of the COVID-19 pandemic has resulted in an unprecedented amount of sequence data of the SARS-CoV-2 genome -- millions of sequences and counting. This amount of data, while being orders of magnitude beyond the capacity of traditional approaches to understanding the diversity, dynamics, and evolution of viruses is nonetheless a rich resource for machine learning (ML) approaches as alternatives for extracting such important information from these data. It is of hence utmost importance to design a framework for testing and benchmarking the robustness of these ML models. This paper makes the first effort (to our knowledge) to benchmark the robustness of ML models by simulating biological sequences with errors. In this paper, we introduce several ways to perturb SARS-CoV-2 genome sequences to mimic the error profiles of common sequencing platforms such as Illumina and PacBio. We show from experiments on a wide array of ML models that some simulation-based approaches are more robust (and accurate) than others for specific embedding methods to certain adversarial attacks to the input sequences. Our benchmarking framework may assist researchers in properly assessing different ML models and help them understand the behavior of the SARS-CoV-2 virus or avoid possible future pandemics.
A k-mer Based Approach for SARS-CoV-2 Variant Identification
Aug 25, 2021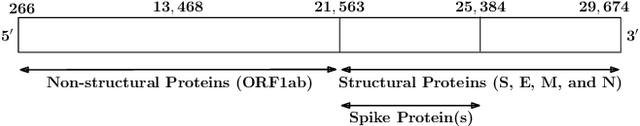
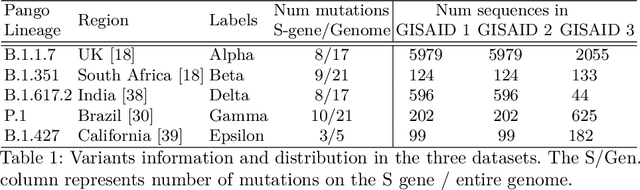
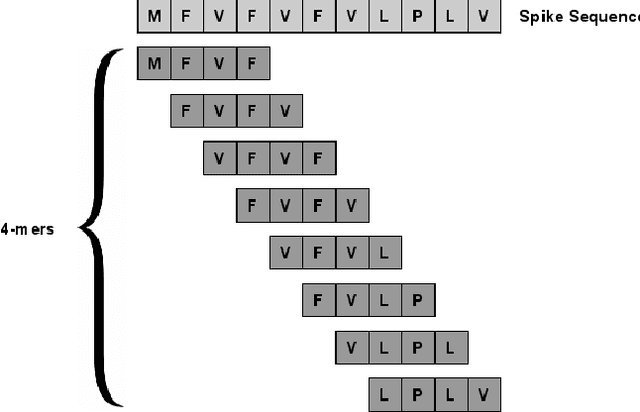
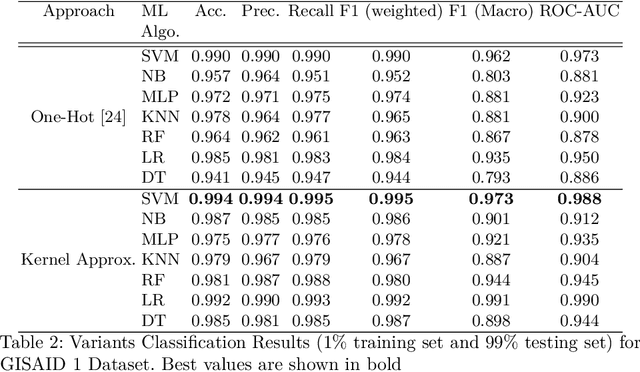
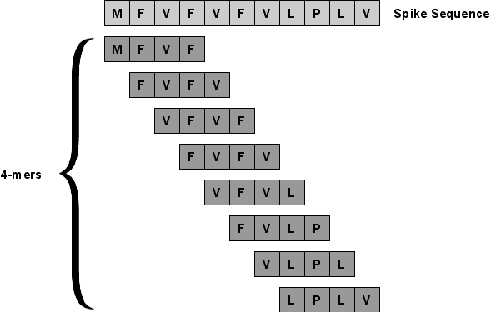
With the rapid spread of the novel coronavirus (COVID-19) across the globe and its continuous mutation, it is of pivotal importance to design a system to identify different known (and unknown) variants of SARS-CoV-2. Identifying particular variants helps to understand and model their spread patterns, design effective mitigation strategies, and prevent future outbreaks. It also plays a crucial role in studying the efficacy of known vaccines against each variant and modeling the likelihood of breakthrough infections. It is well known that the spike protein contains most of the information/variation pertaining to coronavirus variants. In this paper, we use spike sequences to classify different variants of the coronavirus in humans. We show that preserving the order of the amino acids helps the underlying classifiers to achieve better performance. We also show that we can train our model to outperform the baseline algorithms using only a small number of training samples ($1\%$ of the data). Finally, we show the importance of the different amino acids which play a key role in identifying variants and how they coincide with those reported by the USA's Centers for Disease Control and Prevention (CDC).