 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMaterials Learning Algorithms (MALA): Scalable Machine Learning for Electronic Structure Calculations in Large-Scale Atomistic Simulations
Nov 29, 2024



We present the Materials Learning Algorithms (MALA) package, a scalable machine learning framework designed to accelerate density functional theory (DFT) calculations suitable for large-scale atomistic simulations. Using local descriptors of the atomic environment, MALA models efficiently predict key electronic observables, including local density of states, electronic density, density of states, and total energy. The package integrates data sampling, model training and scalable inference into a unified library, while ensuring compatibility with standard DFT and molecular dynamics codes. We demonstrate MALA's capabilities with examples including boron clusters, aluminum across its solid-liquid phase boundary, and predicting the electronic structure of a stacking fault in a large beryllium slab. Scaling analyses reveal MALA's computational efficiency and identify bottlenecks for future optimization. With its ability to model electronic structures at scales far beyond standard DFT, MALA is well suited for modeling complex material systems, making it a versatile tool for advanced materials research.
Accelerating Finite-temperature Kohn-Sham Density Functional Theory with Deep Neural Networks
Oct 10, 2020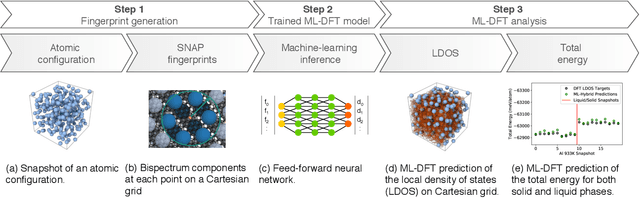

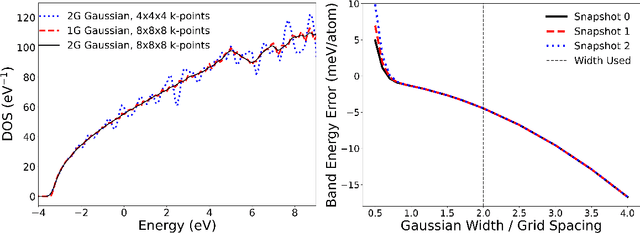
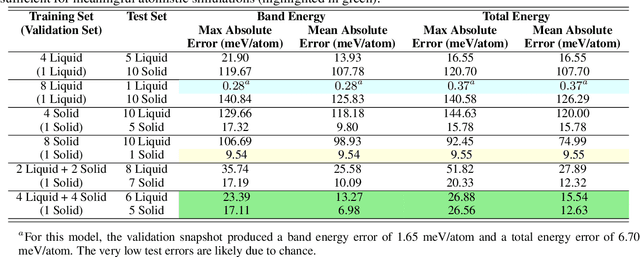
We present a numerical modeling workflow based on machine learning (ML) which reproduces the the total energies produced by Kohn-Sham density functional theory (DFT) at finite electronic temperature to within chemical accuracy at negligible computational cost. Based on deep neural networks, our workflow yields the local density of states (LDOS) for a given atomic configuration. From the LDOS, spatially-resolved, energy-resolved, and integrated quantities can be calculated, including the DFT total free energy, which serves as the Born-Oppenheimer potential energy surface for the atoms. We demonstrate the efficacy of this approach for both solid and liquid metals and compare results between independent and unified machine-learning models for solid and liquid aluminum. Our machine-learning density functional theory framework opens up the path towards multiscale materials modeling for matter under ambient and extreme conditions at a computational scale and cost that is unattainable with current algorithms.
Simple and efficient algorithms for training machine learning potentials to force data
Jun 09, 2020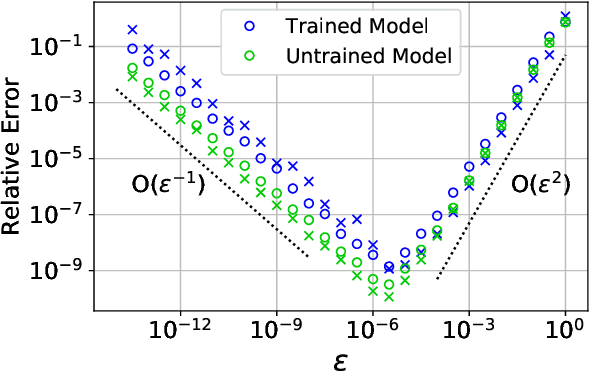
Abstract Machine learning models, trained on data from ab initio quantum simulations, are yielding molecular dynamics potentials with unprecedented accuracy. One limiting factor is the quantity of available training data, which can be expensive to obtain. A quantum simulation often provides all atomic forces, in addition to the total energy of the system. These forces provide much more information than the energy alone. It may appear that training a model to this large quantity of force data would introduce significant computational costs. Actually, training to all available force data should only be a few times more expensive than training to energies alone. Here, we present a new algorithm for efficient force training, and benchmark its accuracy by training to forces from real-world datasets for organic chemistry and bulk aluminum.