 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExploring Gene Regulatory Interaction Networks and predicting therapeutic molecules for Hypopharyngeal Cancer and EGFR-mutated lung adenocarcinoma
Feb 27, 2024With the advent of Information technology, the Bioinformatics research field is becoming increasingly attractive to researchers and academicians. The recent development of various Bioinformatics toolkits has facilitated the rapid processing and analysis of vast quantities of biological data for human perception. Most studies focus on locating two connected diseases and making some observations to construct diverse gene regulatory interaction networks, a forerunner to general drug design for curing illness. For instance, Hypopharyngeal cancer is a disease that is associated with EGFR-mutated lung adenocarcinoma. In this study, we select EGFR-mutated lung adenocarcinoma and Hypopharyngeal cancer by finding the Lung metastases in hypopharyngeal cancer. To conduct this study, we collect Mircorarray datasets from GEO (Gene Expression Omnibus), an online database controlled by NCBI. Differentially expressed genes, common genes, and hub genes between the selected two diseases are detected for the succeeding move. Our research findings have suggested common therapeutic molecules for the selected diseases based on 10 hub genes with the highest interactions according to the degree topology method and the maximum clique centrality (MCC). Our suggested therapeutic molecules will be fruitful for patients with those two diseases simultaneously.
Predicting Patient COVID-19 Disease Severity by means of Statistical and Machine Learning Analysis of Blood Cell Transcriptome Data
Nov 19, 2020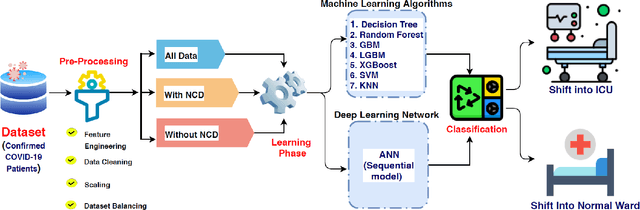
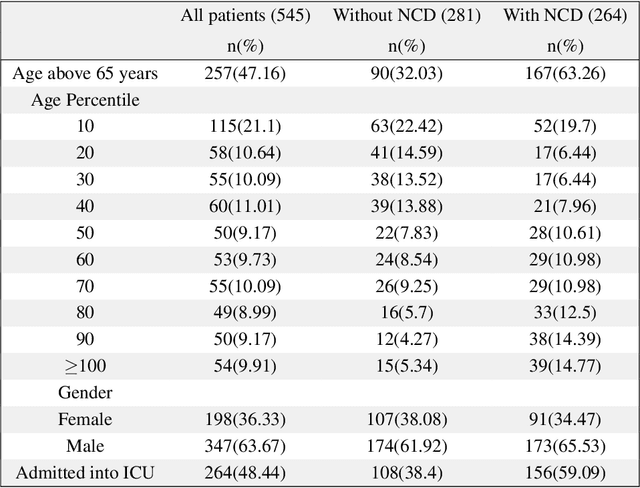
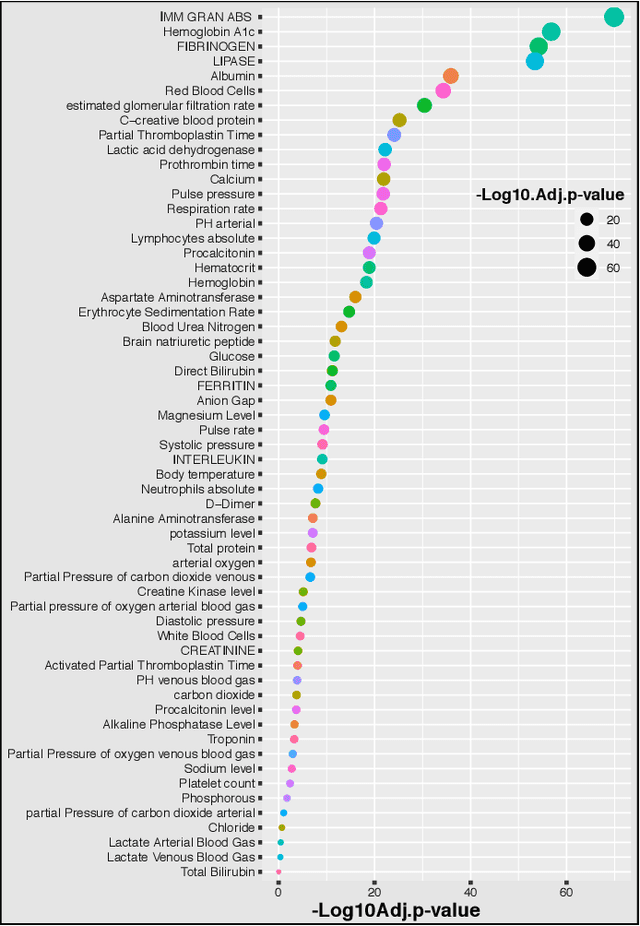
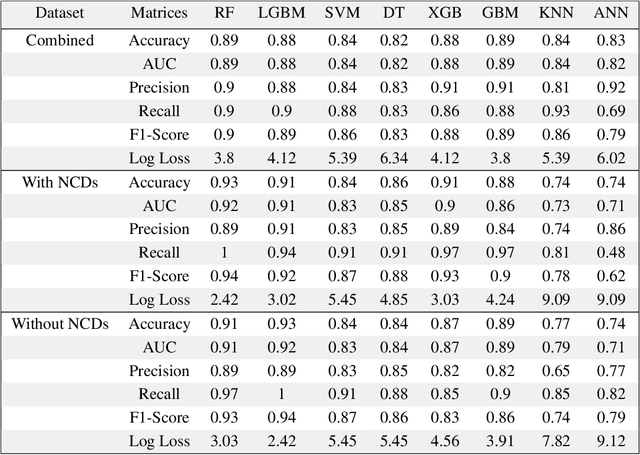
Introduction: For COVID-19 patients accurate prediction of disease severity and mortality risk would greatly improve care delivery and resource allocation. There are many patient-related factors, such as pre-existing comorbidities that affect disease severity. Since rapid automated profiling of peripheral blood samples is widely available, we investigated how such data from the peripheral blood of COVID-19 patients might be used to predict clinical outcomes. Methods: We thus investigated such clinical datasets from COVID-19 patients with known outcomes by combining statistical comparison and correlation methods with machine learning algorithms; the latter included decision tree, random forest, variants of gradient boosting machine, support vector machine, K-nearest neighbour and deep learning methods. Results: Our work revealed several clinical parameters measurable in blood samples, which discriminated between healthy people and COVID-19 positive patients and showed predictive value for later severity of COVID-19 symptoms. We thus developed a number of analytic methods that showed accuracy and precision for disease severity and mortality outcome predictions that were above 90%. Conclusions: In sum, we developed methodologies to analyse patient routine clinical data which enables more accurate prediction of COVID-19 patient outcomes. This type of approaches could, by employing standard hospital laboratory analyses of patient blood, be utilised to identify, COVID-19 patients at high risk of mortality and so enable their treatment to be optimised.