 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSubstructure-Atom Cross Attention for Molecular Representation Learning
Paper and Code

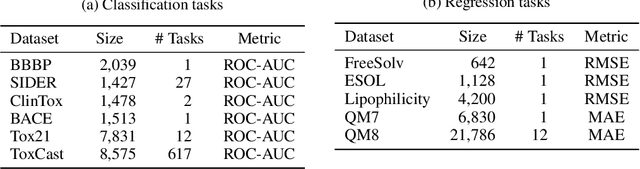
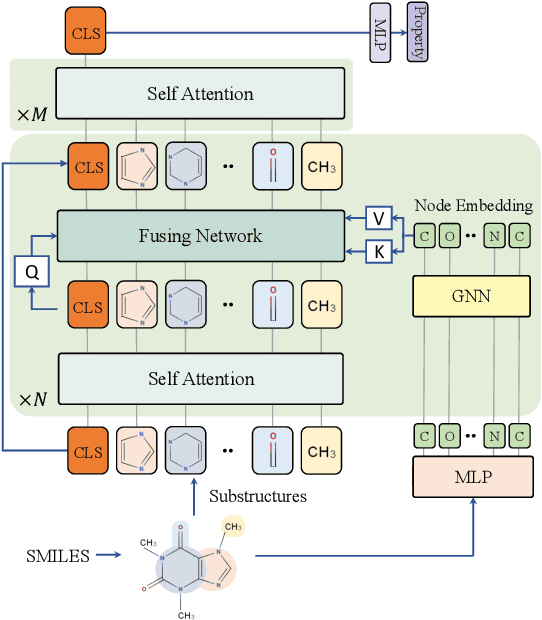
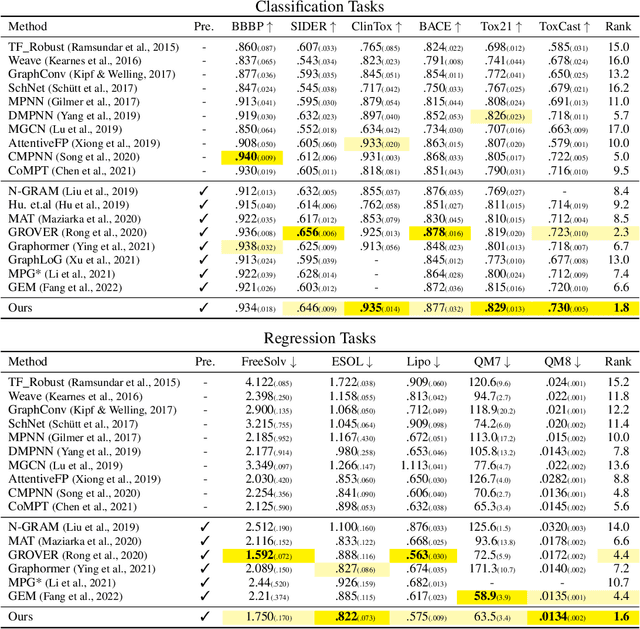
Designing a neural network architecture for molecular representation is crucial for AI-driven drug discovery and molecule design. In this work, we propose a new framework for molecular representation learning. Our contribution is threefold: (a) demonstrating the usefulness of incorporating substructures to node-wise features from molecules, (b) designing two branch networks consisting of a transformer and a graph neural network so that the networks fused with asymmetric attention, and (c) not requiring heuristic features and computationally-expensive information from molecules. Using 1.8 million molecules collected from ChEMBL and PubChem database, we pretrain our network to learn a general representation of molecules with minimal supervision. The experimental results show that our pretrained network achieves competitive performance on 11 downstream tasks for molecular property prediction.