 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning Graph Models for Template-Free Retrosynthesis
Paper and Code
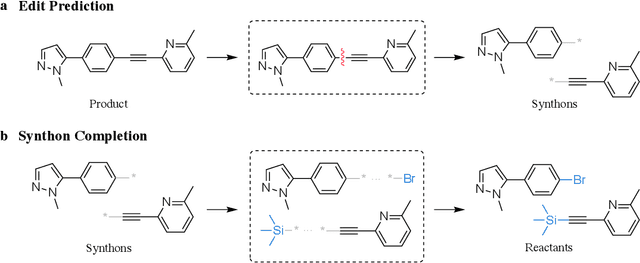
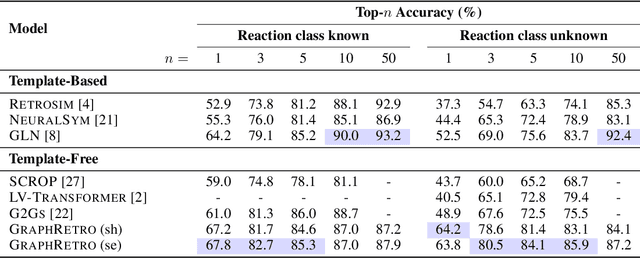
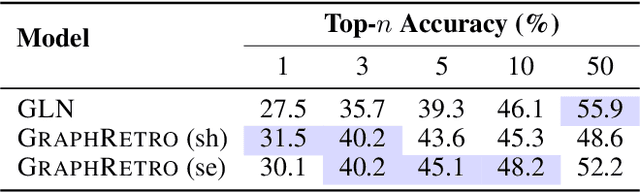
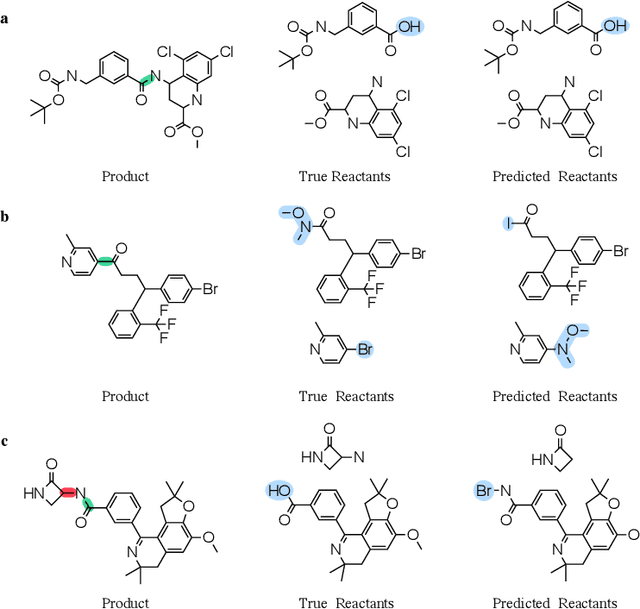
Retrosynthesis prediction is a fundamental problem in organic synthesis, where the task is to identify precursor molecules that can be used to synthesize a target molecule. Despite recent advancements in neural retrosynthesis algorithms, they are unable to fully recapitulate the strategies employed by chemists and do not generalize well to infrequent reaction types. In this paper, we propose a graph-based approach that capitalizes on the idea that the graph topology of precursor molecules is largely unaltered during the reaction. The model first predicts the set of graph edits transforming the target into incomplete molecules called synthons. Next, the model learns to expand synthons into complete molecules by attaching relevant leaving groups. Since the model operates at the level of molecular fragments, it avoids full generation, greatly simplifying the underlying architecture and improving its ability to generalize. The model yields $11.7\%$ absolute improvement over state-of-the-art approaches on the USPTO-50k dataset, and a $4\%$ absolute improvement on a rare reaction subset of the same dataset.