 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Polish: A Novel Graph Generation Paradigm for Molecular Optimization
Paper and Code
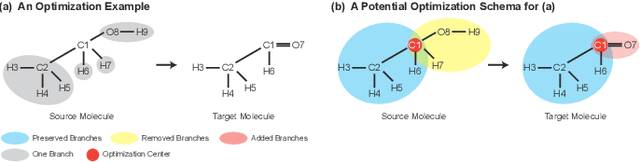
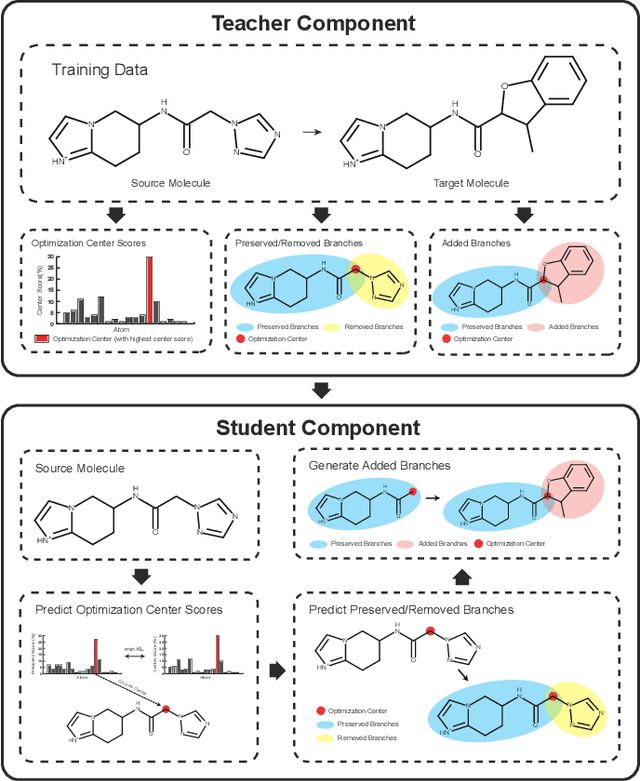
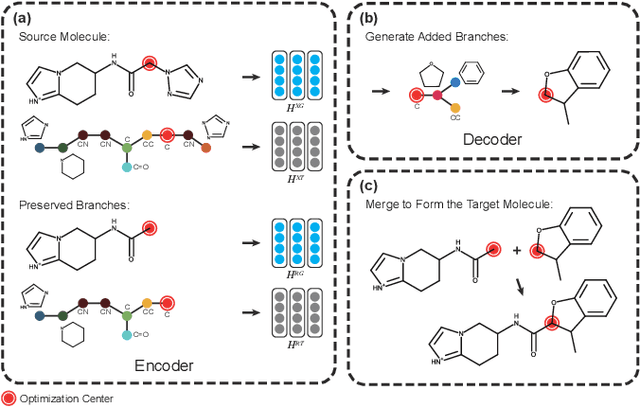
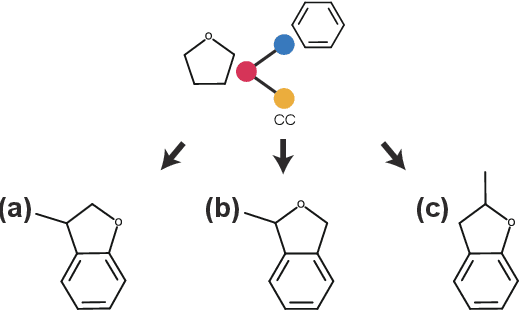
Molecular optimization, which transforms a given input molecule X into another Y with desirable properties, is essential in molecular drug discovery. The traditional translating approaches, generating the molecular graphs from scratch by adding some substructures piece by piece, prone to error because of the large set of candidate substructures in a large number of steps to the final target. In this study, we present a novel molecular optimization paradigm, Graph Polish, which changes molecular optimization from the traditional "two-language translating" task into a "single-language polishing" task. The key to this optimization paradigm is to find an optimization center subject to the conditions that the preserved areas around it ought to be maximized and thereafter the removed and added regions should be minimized. We then propose an effective and efficient learning framework T&S polish to capture the long-term dependencies in the optimization steps. The T component automatically identifies and annotates the optimization centers and the preservation, removal and addition of some parts of the molecule, and the S component learns these behaviors and applies these actions to a new molecule. Furthermore, the proposed paradigm can offer an intuitive interpretation for each molecular optimization result. Experiments with multiple optimization tasks are conducted on four benchmark datasets. The proposed T&S polish approach achieves significant advantage over the five state-of-the-art baseline methods on all the tasks. In addition, extensive studies are conducted to validate the effectiveness, explainability and time saving of the novel optimization paradigm.