 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph-based Molecular Representation Learning
Paper and Code


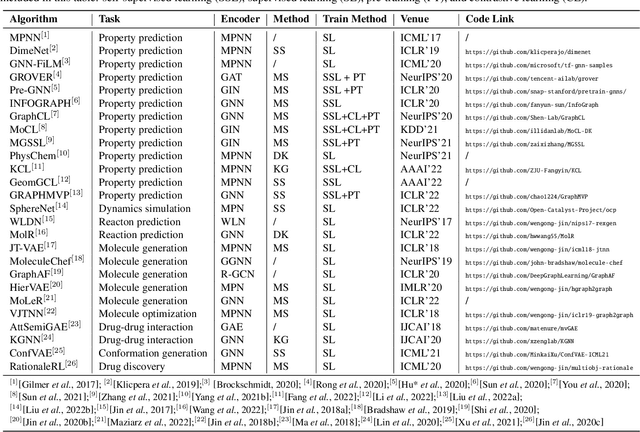
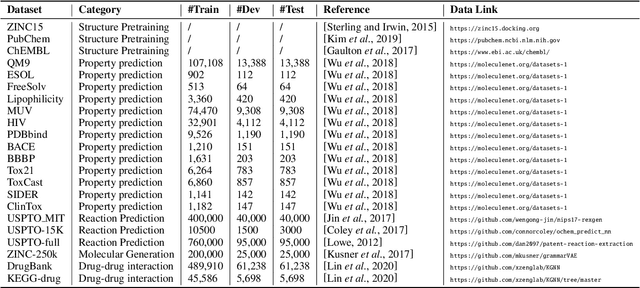
Molecular representation learning (MRL) is a key step to build the connection between machine learning and chemical science. In particular, it encodes molecules as numerical vectors preserving the molecular structures and features, on top of which the downstream tasks (e.g., property prediction) can be performed. Recently, MRL has achieved considerable progress, especially in deep molecular graph learning-based methods. In this survey, we systematically review these graph-based molecular representation techniques. Specifically, we first introduce the data and features of the 2D and 3D graph molecular datasets. Then we summarize the methods specially designed for MRL and categorize them into four strategies. Furthermore, we discuss some typical chemical applications supported by MRL. To facilitate studies in this fast-developing area, we also list the benchmarks and commonly used datasets in the paper. Finally, we share our thoughts on future research directions.