 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChemBO: Bayesian Optimization of Small Organic Molecules with Synthesizable Recommendations
Paper and Code
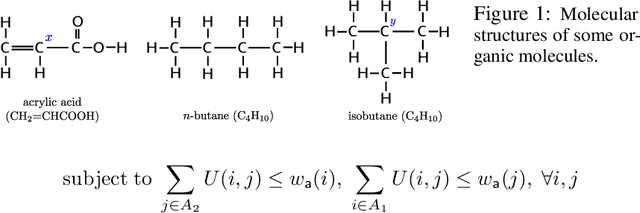

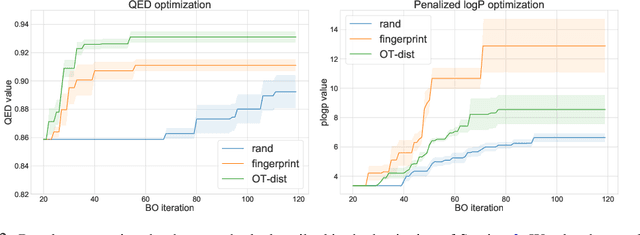
We describe ChemBO, a Bayesian Optimization framework for generating and optimizing organic molecules for desired molecular properties. This framework is useful in applications such as drug discovery, where an algorithm recommends new candidate molecules; these molecules first need to be synthesized and then tested for drug-like properties. The algorithm uses the results of past tests to recommend new ones so as to find good molecules efficiently. Most existing data-driven methods for this problem do not account for sample efficiency and/or fail to enforce realistic constraints on synthesizability. In this work, we explore existing kernels for molecules in the literature as well as propose a novel kernel which views a molecule as a graph. In ChemBO, we implement these kernels in a Gaussian process model. Then we explore the chemical space by traversing possible paths of molecular synthesis. Consequently, our approach provides a proposal synthesis path every time it recommends a new molecule to test, a crucial advantage when compared to existing methods. In our experiments, we demonstrate the efficacy of the proposed approach on several molecular optimization problems.