 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBatmanNet: Bi-branch Masked Graph Transformer Autoencoder for Molecular Representation
Paper and Code
Nov 29, 2022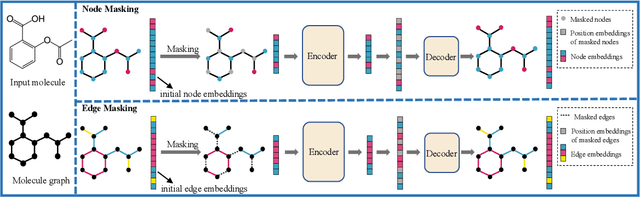
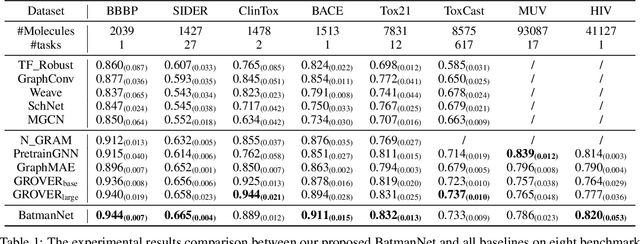
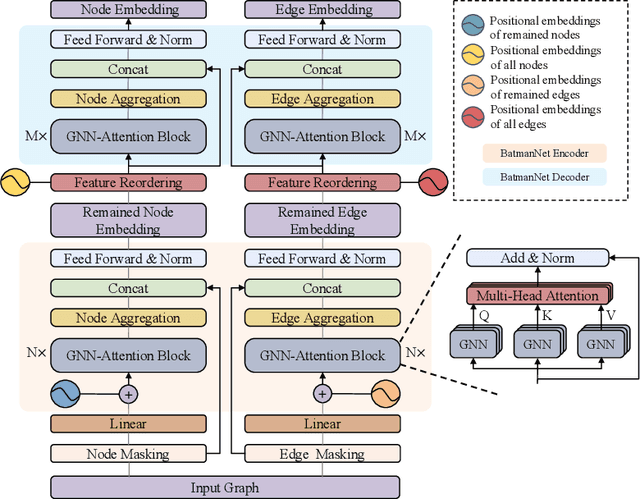
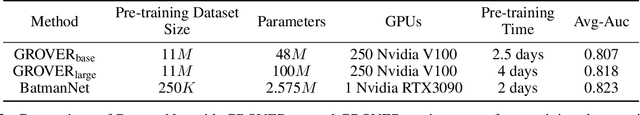
Although substantial efforts have been made using graph neural networks (GNNs) for AI-driven drug discovery (AIDD), effective molecular representation learning remains an open challenge, especially in the case of insufficient labeled molecules. Recent studies suggest that big GNN models pre-trained by self-supervised learning on unlabeled datasets enable better transfer performance in downstream molecular property prediction tasks. However, they often require large-scale datasets and considerable computational resources, which is time-consuming, computationally expensive, and environmentally unfriendly. To alleviate these limitations, we propose a novel pre-training model for molecular representation learning, Bi-branch Masked Graph Transformer Autoencoder (BatmanNet). BatmanNet features two tailored and complementary graph autoencoders to reconstruct the missing nodes and edges from a masked molecular graph. To our surprise, BatmanNet discovered that the highly masked proportion (60%) of the atoms and bonds achieved the best performance. We further propose an asymmetric graph-based encoder-decoder architecture for either nodes and edges, where a transformer-based encoder only takes the visible subset of nodes or edges, and a lightweight decoder reconstructs the original molecule from the latent representation and mask tokens. With this simple yet effective asymmetrical design, our BatmanNet can learn efficiently even from a much smaller-scale unlabeled molecular dataset to capture the underlying structural and semantic information, overcoming a major limitation of current deep neural networks for molecular representation learning. For instance, using only 250K unlabelled molecules as pre-training data, our BatmanNet with 2.575M parameters achieves a 0.5% improvement on the average AUC compared with the current state-of-the-art method with 100M parameters pre-trained on 11M molecules.