 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNearly Optimal Bayesian Inference for Structural Missingness
Jan 27, 2026Structural missingness breaks 'just impute and train': values can be undefined by causal or logical constraints, and the mask may depend on observed variables, unobserved variables (MNAR), and other missingness indicators. It simultaneously brings (i) a catch-22 situation with causal loop, prediction needs the missing features, yet inferring them depends on the missingness mechanism, (ii) under MNAR, the unseen are different, the missing part can come from a shifted distribution, and (iii) plug-in imputation, a single fill-in can lock in uncertainty and yield overconfident, biased decisions. In the Bayesian view, prediction via the posterior predictive distribution integrates over the full model posterior uncertainty, rather than relying on a single point estimate. This framework decouples (i) learning an in-model missing-value posterior from (ii) label prediction by optimizing the predictive posterior distribution, enabling posterior integration. This decoupling yields an in-model almost-free-lunch: once the posterior is learned, prediction is plug-and-play while preserving uncertainty propagation. It achieves SOTA on 43 classification and 15 imputation benchmarks, with finite-sample near Bayes-optimality guarantees under our SCM prior.
DGoT: Dynamic Graph of Thoughts for Scientific Abstract Generation
Mar 26, 2024The method of training language models based on domain datasets has obtained significant achievements in the task of generating scientific paper abstracts. However, such models face problems of generalization and expensive training costs. The use of large language models (LLMs) to solve the task of generating paper abstracts saves the cost of model training. However, due to the hallucination problem of LLM, it is often necessary to improve the reliability of the results through multi-round query prompt approach such as Graph of Thoughts (GoT), which also brings additional reasoning costs. In this paper, we propose a Dynamic Graph of Thought (DGoT). It not only inherits the advantages of the existing GoT prompt approach, but also dynamically adjust the graph structure according to data characteristics while reducing model reasoning cost. Experimental results show that our method's cost-effectiveness in abstract generation tasks is only 43.7% to 56.4% of other multi-round query prompt approaches. Our code is available at https://github.com/JayceNing/DGoT.
Schrödinger-ANI: An Eight-Element Neural Network Interaction Potential with Greatly Expanded Coverage of Druglike Chemical Space
Nov 22, 2019
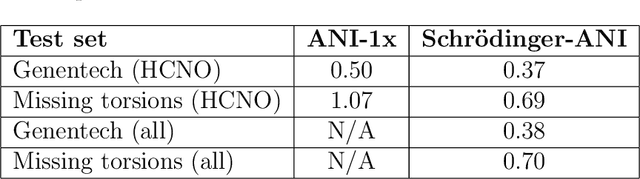
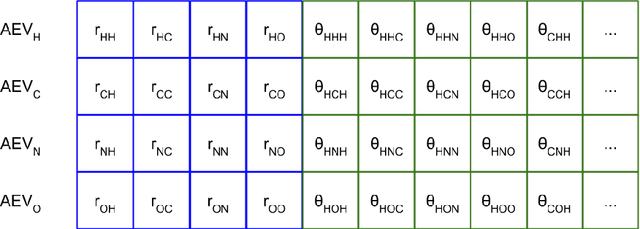
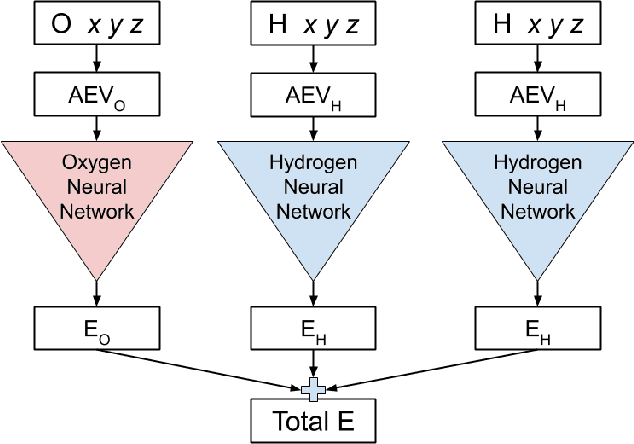
We have developed a neural network potential energy function for use in drug discovery, with chemical element support extended from 41% to 94% of druglike molecules based on ChEMBL. We expand on the work of Smith et al., with their highly accurate network for the elements H, C, N, O, creating a network for H, C, N, O, S, F, Cl, P. We focus particularly on the calculation of relative conformer energies, for which we show that our new potential energy function has an RMSE of 0.70 kcal/mol for prospective druglike molecule conformers, substantially better than the previous state of the art. The speed and accuracy of this model could greatly accelerate the parameterization of protein-ligand binding free energy calculations for novel druglike molecules.