 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSchrödinger-ANI: An Eight-Element Neural Network Interaction Potential with Greatly Expanded Coverage of Druglike Chemical Space
Nov 22, 2019
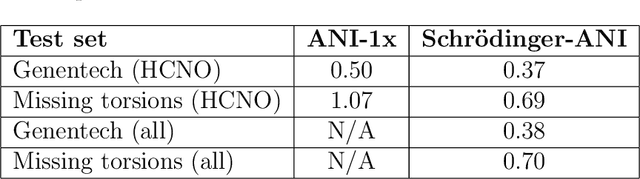
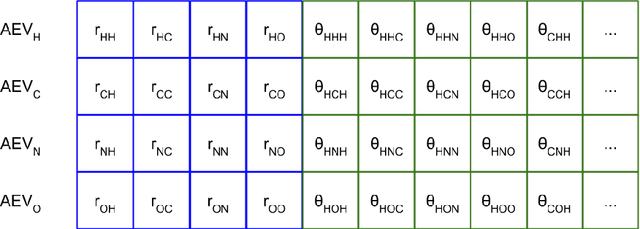
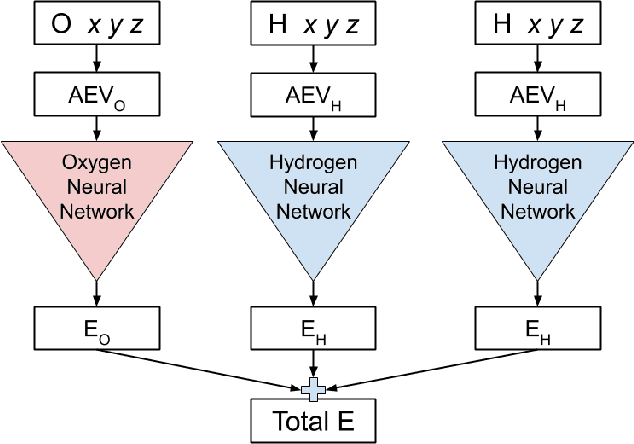
We have developed a neural network potential energy function for use in drug discovery, with chemical element support extended from 41% to 94% of druglike molecules based on ChEMBL. We expand on the work of Smith et al., with their highly accurate network for the elements H, C, N, O, creating a network for H, C, N, O, S, F, Cl, P. We focus particularly on the calculation of relative conformer energies, for which we show that our new potential energy function has an RMSE of 0.70 kcal/mol for prospective druglike molecule conformers, substantially better than the previous state of the art. The speed and accuracy of this model could greatly accelerate the parameterization of protein-ligand binding free energy calculations for novel druglike molecules.
Molecular Structure Extraction From Documents Using Deep Learning
Feb 14, 2018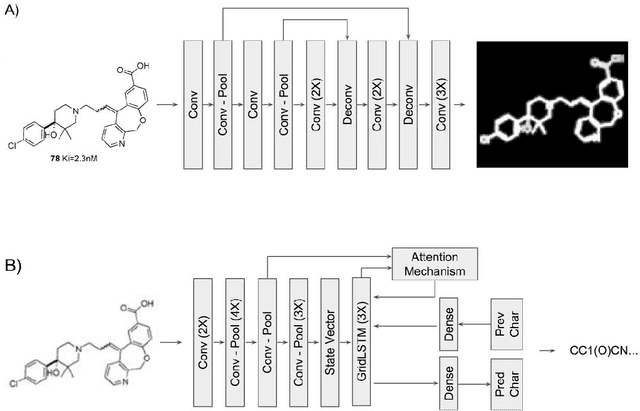

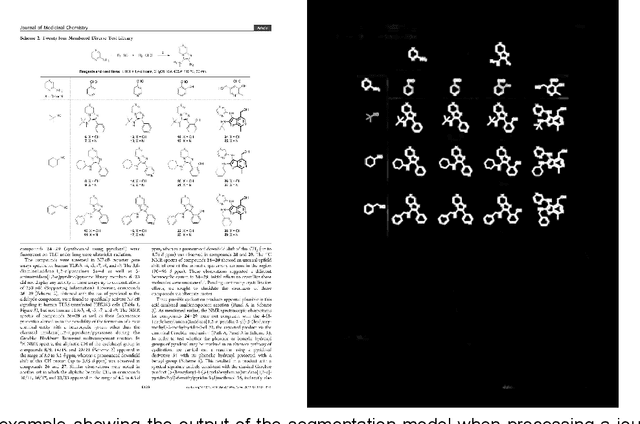
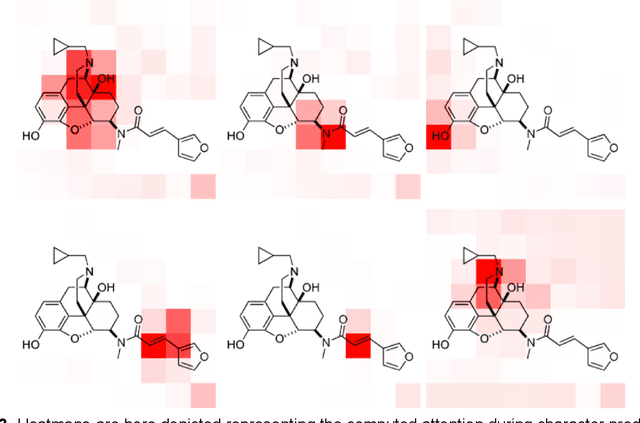
Chemical structure extraction from documents remains a hard problem due to both false positive identification of structures during segmentation and errors in the predicted structures. Current approaches rely on handcrafted rules and subroutines that perform reasonably well generally, but still routinely encounter situations where recognition rates are not yet satisfactory and systematic improvement is challenging. Complications impacting performance of current approaches include the diversity in visual styles used by various software to render structures, the frequent use of ad hoc annotations, and other challenges related to image quality, including resolution and noise. We here present end-to-end deep learning solutions for both segmenting molecular structures from documents and for predicting chemical structures from these segmented images. This deep learning-based approach does not require any handcrafted features, is learned directly from data, and is robust against variations in image quality and style. Using the deep-learning approach described herein we show that it is possible to perform well on both segmentation and prediction of low resolution images containing moderately sized molecules found in journal articles and patents.