 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtein Classification using Machine Learning and Statistical Techniques: A Comparative Analysis
Jan 18, 2019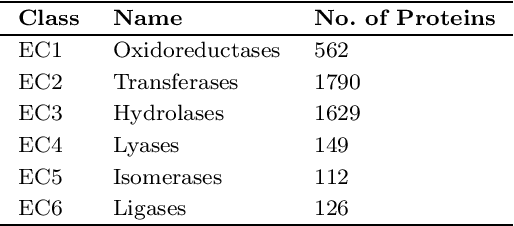
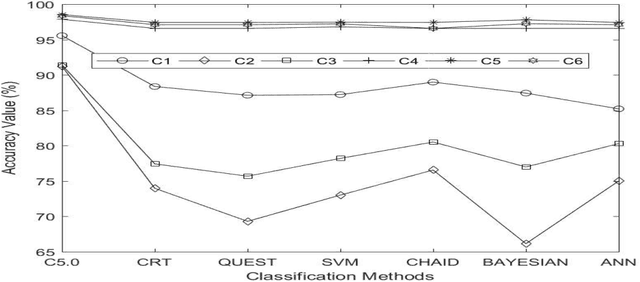

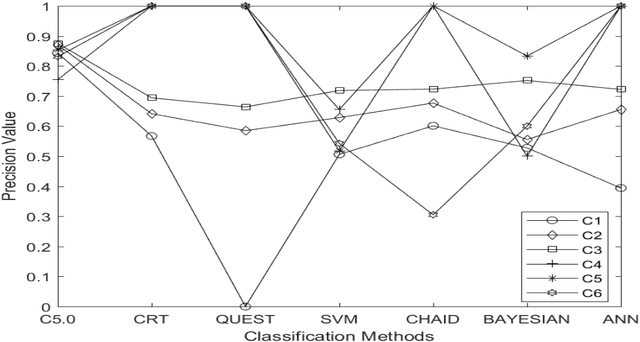
In recent era prediction of enzyme class from an unknown protein is one of the challenging tasks in bioinformatics. Day to day the number of proteins is increases as result the prediction of enzyme class gives a new opportunity to bioinformatics scholars. The prime objective of this article is to implement the machine learning classification technique for feature selection and predictions also find out an appropriate classification technique for function prediction. In this article the seven different classification technique like CRT, QUEST, CHAID, C5.0, ANN (Artificial Neural Network), SVM and Bayesian has been implemented on 4368 protein data that has been extracted from UniprotKB databank and categories into six different class. The proteins data is high dimensional sequence data and contain a maximum of 48 features.To manipulate the high dimensional sequential protein data with different classification technique, the SPSS has been used as an experimental tool. Different classification techniques give different results for every model and shows that the data are imbalanced for class C4, C5 and C6. The imbalanced data affect the performance of model. In these three classes the precision and recall value is very less or negligible. The experimental results highlight that the C5.0 classification technique accuracy is more suited for protein feature classification and predictions. The C5.0 classification technique gives 95.56% accuracy and also gives high precision and recall value. Finally, we conclude that the features that is selected can be used for function prediction.
Deep Robust Framework for Protein Function Prediction using Variable-Length Protein Sequences
Nov 04, 2018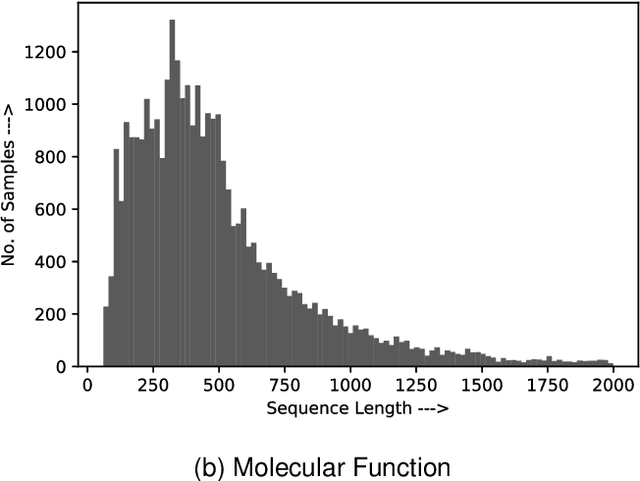
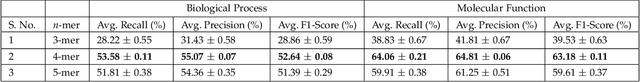
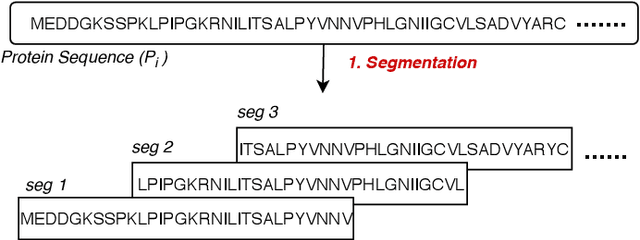
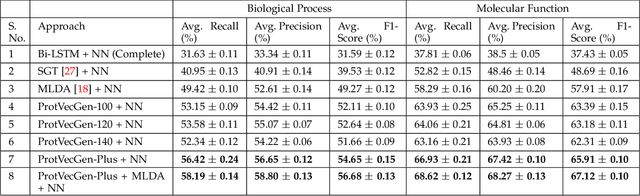
Amino acid sequence portrays most intrinsic form of a protein and expresses primary structure of protein. The order of amino acids in a sequence enables a protein to acquire a particular stable conformation that is responsible for the functions of the protein. This relationship between a sequence and its function motivates the need to analyse the sequences for predicting protein functions. Early generation computational methods using BLAST, FASTA, etc. perform function transfer based on sequence similarity with existing databases and are computationally slow. Although machine learning based approaches are fast, they fail to perform well for long protein sequences (i.e., protein sequences with more than 300 amino acid residues). In this paper, we introduce a novel method for construction of two separate feature sets for protein sequences based on analysis of 1) single fixed-sized segments and 2) multi-sized segments, using bi-directional long short-term memory network. Further, model based on proposed feature set is combined with the state of the art Multi-lable Linear Discriminant Analysis (MLDA) features based model to improve the accuracy. Extensive evaluations using separate datasets for biological processes and molecular functions demonstrate promising results for both single-sized and multi-sized segments based feature sets. While former showed an improvement of +3.37% and +5.48%, the latter produces an improvement of +5.38% and +8.00% respectively for two datasets over the state of the art MLDA based classifier. After combining two models, there is a significant improvement of +7.41% and +9.21% respectively for two datasets compared to MLDA based classifier. Specifically, the proposed approach performed well for the long protein sequences and superior overall performance.