 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStutterFuse: Mitigating Modality Collapse in Stuttering Detection with Jaccard-Weighted Metric Learning and Gated Fusion
Dec 15, 2025Stuttering detection breaks down when disfluencies overlap. Existing parametric models struggle to distinguish complex, simultaneous disfluencies (e.g., a 'block' with a 'prolongation') due to the scarcity of these specific combinations in training data. While Retrieval-Augmented Generation (RAG) has revolutionized NLP by grounding models in external knowledge, this paradigm remains unexplored in pathological speech processing. To bridge this gap, we introduce StutterFuse, the first Retrieval-Augmented Classifier (RAC) for multi-label stuttering detection. By conditioning a Conformer encoder on a non-parametric memory bank of clinical examples, we allow the model to classify by reference rather than memorization. We further identify and solve "Modality Collapse", an "Echo Chamber" effect where naive retrieval boosts recall but degrades precision. We mitigate this using: (1) SetCon, a Jaccard-Weighted Metric Learning objective that optimizes for multi-label set similarity, and (2) a Gated Mixture-of-Experts fusion strategy that dynamically arbitrates between acoustic evidence and retrieved context. On the SEP-28k dataset, StutterFuse achieves a weighted F1-score of 0.65, outperforming strong baselines and demonstrating remarkable zero-shot cross-lingual generalization.
λ-Scaled-Attention: A Novel Fast Attention Mechanism for Efficient Modeling of Protein Sequences
Jan 09, 2022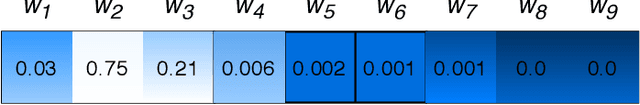
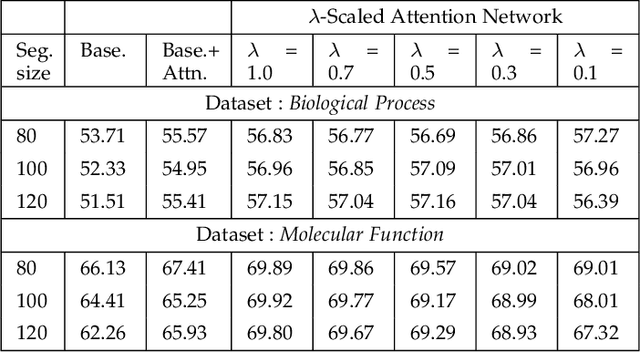
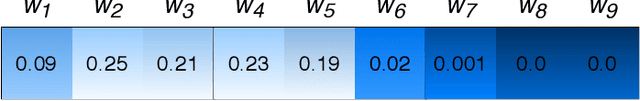
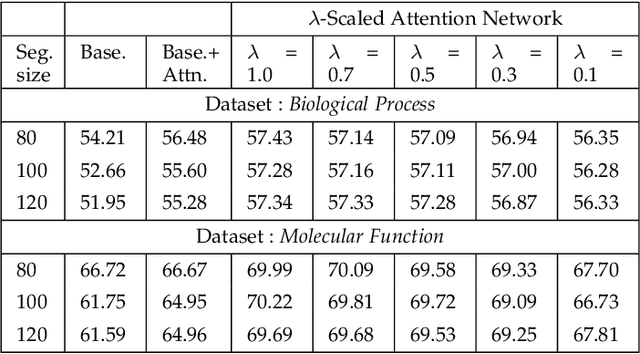
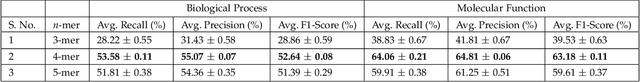
Attention-based deep networks have been successfully applied on textual data in the field of NLP. However, their application on protein sequences poses additional challenges due to the weak semantics of the protein words, unlike the plain text words. These unexplored challenges faced by the standard attention technique include (i) vanishing attention score problem and (ii) high variations in the attention distribution. In this regard, we introduce a novel {\lambda}-scaled attention technique for fast and efficient modeling of the protein sequences that addresses both the above problems. This is used to develop the {\lambda}-scaled attention network and is evaluated for the task of protein function prediction implemented at the protein sub-sequence level. Experiments on the datasets for biological process (BP) and molecular function (MF) showed significant improvements in the F1 score values for the proposed {\lambda}-scaled attention technique over its counterpart approach based on the standard attention technique (+2.01% for BP and +4.67% for MF) and state-of-the-art ProtVecGen-Plus approach (+2.61% for BP and +4.20% for MF). Further, fast convergence (converging in half the number of epochs) and efficient learning (in terms of very low difference between the training and validation losses) were also observed during the training process.
Deep Robust Framework for Protein Function Prediction using Variable-Length Protein Sequences
Nov 04, 2018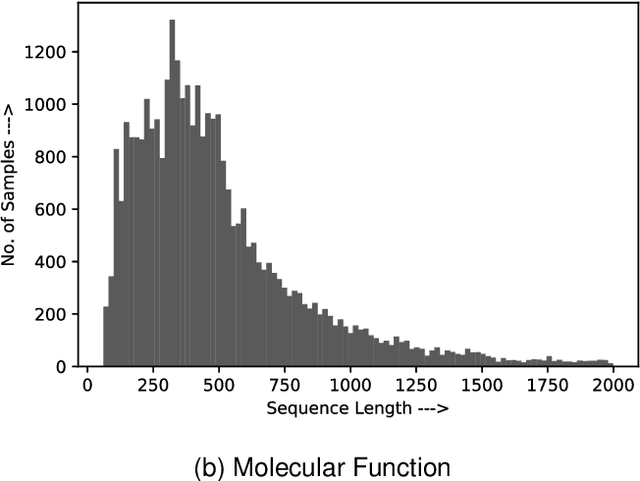
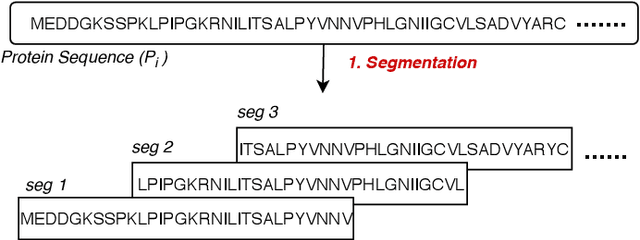
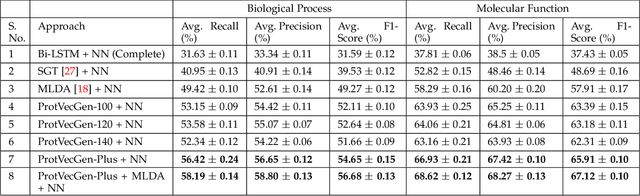
Amino acid sequence portrays most intrinsic form of a protein and expresses primary structure of protein. The order of amino acids in a sequence enables a protein to acquire a particular stable conformation that is responsible for the functions of the protein. This relationship between a sequence and its function motivates the need to analyse the sequences for predicting protein functions. Early generation computational methods using BLAST, FASTA, etc. perform function transfer based on sequence similarity with existing databases and are computationally slow. Although machine learning based approaches are fast, they fail to perform well for long protein sequences (i.e., protein sequences with more than 300 amino acid residues). In this paper, we introduce a novel method for construction of two separate feature sets for protein sequences based on analysis of 1) single fixed-sized segments and 2) multi-sized segments, using bi-directional long short-term memory network. Further, model based on proposed feature set is combined with the state of the art Multi-lable Linear Discriminant Analysis (MLDA) features based model to improve the accuracy. Extensive evaluations using separate datasets for biological processes and molecular functions demonstrate promising results for both single-sized and multi-sized segments based feature sets. While former showed an improvement of +3.37% and +5.48%, the latter produces an improvement of +5.38% and +8.00% respectively for two datasets over the state of the art MLDA based classifier. After combining two models, there is a significant improvement of +7.41% and +9.21% respectively for two datasets compared to MLDA based classifier. Specifically, the proposed approach performed well for the long protein sequences and superior overall performance.