 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReCasNet: Improving consistency within the two-stage mitosis detection framework
Feb 28, 2022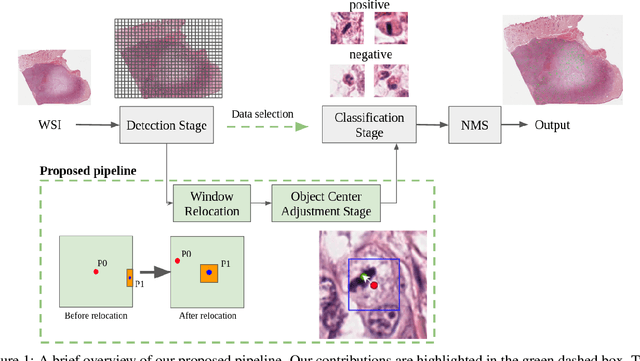
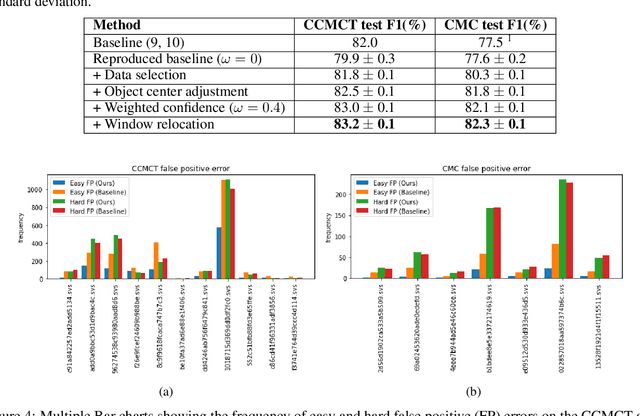
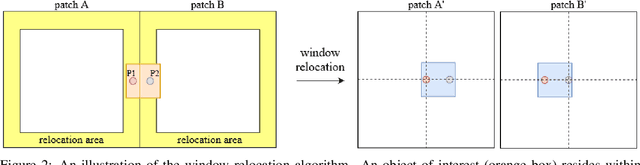

Mitotic count (MC) is an important histological parameter for cancer diagnosis and grading, but the manual process for obtaining MC from whole-slide histopathological images is very time-consuming and prone to error. Therefore, deep learning models have been proposed to facilitate this process. Existing approaches utilize a two-stage pipeline: the detection stage for identifying the locations of potential mitotic cells and the classification stage for refining prediction confidences. However, this pipeline formulation can lead to inconsistencies in the classification stage due to the poor prediction quality of the detection stage and the mismatches in training data distributions between the two stages. In this study, we propose a Refine Cascade Network (ReCasNet), an enhanced deep learning pipeline that mitigates the aforementioned problems with three improvements. First, window relocation was used to reduce the number of poor quality false positives generated during the detection stage. Second, object re-cropping was performed with another deep learning model to adjust poorly centered objects. Third, improved data selection strategies were introduced during the classification stage to reduce the mismatches in training data distributions. ReCasNet was evaluated on two large-scale mitotic figure recognition datasets, canine cutaneous mast cell tumor (CCMCT) and canine mammary carcinoma (CMC), which resulted in up to 4.8% percentage point improvements in the F1 scores for mitotic cell detection and 44.1% reductions in mean absolute percentage error (MAPE) for MC prediction. Techniques that underlie ReCasNet can be generalized to other two-stage object detection networks and should contribute to improving the performances of deep learning models in broad digital pathology applications.
High resolution weakly supervised localization architectures for medical images
Oct 22, 2020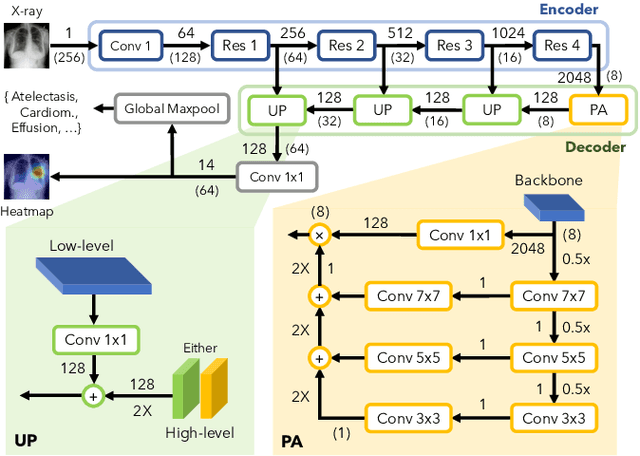
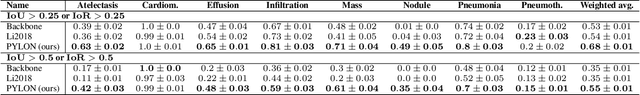
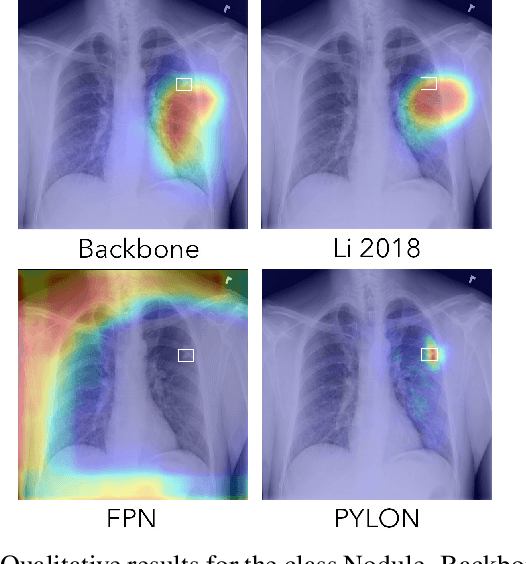
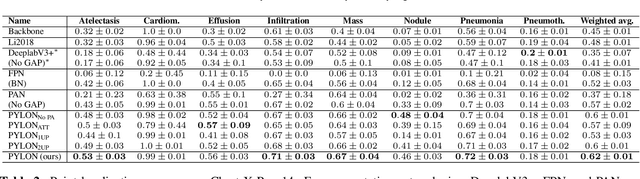
In medical imaging, Class-Activation Map (CAM) serves as the main explainability tool by pointing to the region of interest. Since the localization accuracy from CAM is constrained by the resolution of the model's feature map, one may expect that segmentation models, which generally have large feature maps, would produce more accurate CAMs. However, we have found that this is not the case due to task mismatch. While segmentation models are developed for datasets with pixel-level annotation, only image-level annotation is available in most medical imaging datasets. Our experiments suggest that Global Average Pooling (GAP) and Group Normalization are the main culprits that worsen the localization accuracy of CAM. To address this issue, we propose Pyramid Localization Network (PYLON), a model for high-accuracy weakly-supervised localization that achieved 0.62 average point localization accuracy on NIH's Chest X-Ray 14 dataset, compared to 0.45 for a traditional CAM model. Source code and extended results are available at https://github.com/cmb-chula/pylon.