 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNeuralPDR: Neural Differential Equations as surrogate models for Photodissociation Regions
Jun 17, 2025Computational astrochemical models are essential for helping us interpret and understand the observations of different astrophysical environments. In the age of high-resolution telescopes such as JWST and ALMA, the substructure of many objects can be resolved, raising the need for astrochemical modeling at these smaller scales, meaning that the simulations of these objects need to include both the physics and chemistry to accurately model the observations. The computational cost of the simulations coupling both the three-dimensional hydrodynamics and chemistry is enormous, creating an opportunity for surrogate models that can effectively substitute the chemical solver. In this work we present surrogate models that can replace the original chemical code, namely Latent Augmented Neural Ordinary Differential Equations. We train these surrogate architectures on three datasets of increasing physical complexity, with the last dataset derived directly from a three-dimensional simulation of a molecular cloud using a Photodissociation Region (PDR) code, 3D-PDR. We show that these surrogate models can provide speedup and reproduce the original observable column density maps of the dataset. This enables the rapid inference of the chemistry (on the GPU), allowing for the faster statistical inference of observations or increasing the resolution in hydrodynamical simulations of astrophysical environments.
Understanding molecular ratios in the carbon and oxygen poor outer Milky Way with interpretable machine learning
May 13, 2025Context. The outer Milky Way has a lower metallicity than our solar neighbourhood, but still many molecules are detected in the region. Molecular line ratios can serve as probes to better understand the chemistry and physics in these regions. Aims. We use interpretable machine learning to study 9 different molecular ratios, helping us understand the forward connection between the physics of these environments and the carbon and oxygen chemistries. Methods. Using a large grid of astrochemical models generated using UCLCHEM, we study the properties of molecular clouds of low oxygen and carbon initial abundance. We first try to understand the line ratios using a classical analysis. We then move on to using interpretable machine learning, namely Shapley Additive Explanations (SHAP), to understand the higher order dependencies of the ratios over the entire parameter grid. Lastly we use the Uniform Manifold Approximation and Projection technique (UMAP) as a reduction method to create intuitive groupings of models. Results. We find that the parameter space is well covered by the line ratios, allowing us to investigate all input parameters. SHAP analysis shows that the temperature and density are the most important features, but the carbon and oxygen abundances are important in parts of the parameter space. Lastly, we find that we can group different types of ratios using UMAP. Conclusions. We show the chosen ratios are mostly sensitive to changes in the carbon initial abundance, together with the temperature and density. Especially the CN/HCN and HNC/HCN ratio are shown to be sensitive to the initial carbon abundance, making them excellent probes for this parameter. Out of the ratios, only CS/SO shows a sensitivity to the oxygen abundance.
3D-PDR Orion dataset and NeuralPDR: Neural Differential Equations for Photodissociation Regions
Dec 01, 2024



We present a novel dataset of simulations of the photodissociation region (PDR) in the Orion Bar and provide benchmarks of emulators for the dataset. Numerical models of PDRs are computationally expensive since the modeling of these changing regions requires resolving the thermal balance and chemical composition along a line-of-sight into an interstellar cloud. This often makes it a bottleneck for 3D simulations of these regions. In this work, we provide a dataset of 8192 models with different initial conditions simulated with 3D-PDR. We then benchmark different architectures, focusing on Augmented Neural Ordinary Differential Equation (ANODE) based models (Code be found at https://github.com/uclchem/neuralpdr). Obtaining fast and robust emulators that can be included as preconditioners of classical codes or full emulators into 3D simulations of PDRs.
Disentangled Representation Learning for Astronomical Chemical Tagging
Mar 10, 2021

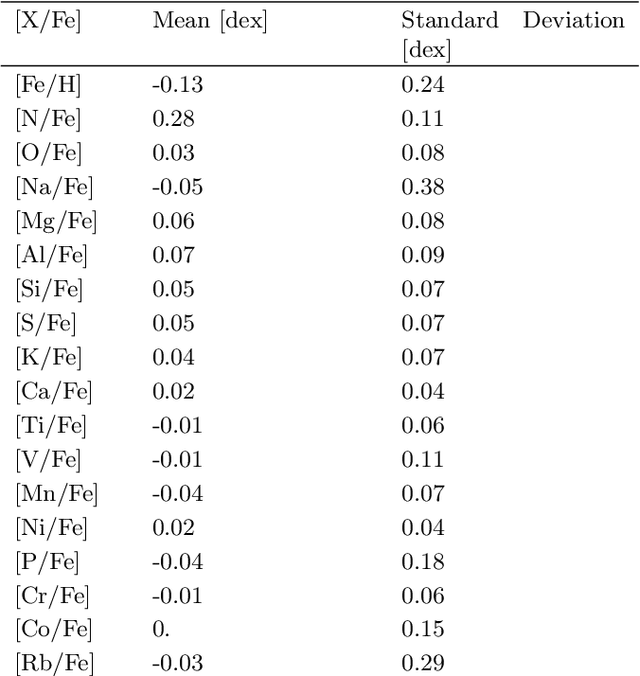
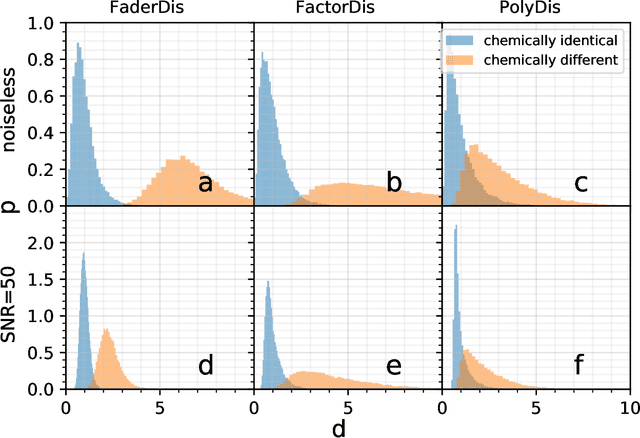
Modern astronomical surveys are observing spectral data for millions of stars. These spectra contain chemical information that can be used to trace the Galaxy's formation and chemical enrichment history. However, extracting the information from spectra, and making precise and accurate chemical abundance measurements are challenging. Here, we present a data-driven method for isolating the chemical factors of variation in stellar spectra from those of other parameters (i.e. \teff, \logg, \feh). This enables us to build a spectral projection for each star with these parameters removed. We do this with no ab initio knowledge of elemental abundances themselves, and hence bypass the uncertainties and systematics associated with modeling that rely on synthetic stellar spectra. To remove known non-chemical factors of variation, we develop and implement a neural network architecture that learns a disentangled spectral representation. We simulate our recovery of chemically identical stars using the disentangled spectra in a synthetic APOGEE-like dataset. We show that this recovery declines as a function of the signal to noise ratio, but that our neural network architecture outperforms simpler modeling choices. Our work demonstrates the feasibility of data-driven abundance-free chemical tagging.