 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInteger linear programming for unsupervised training set selection in molecular machine learning
Oct 21, 2024
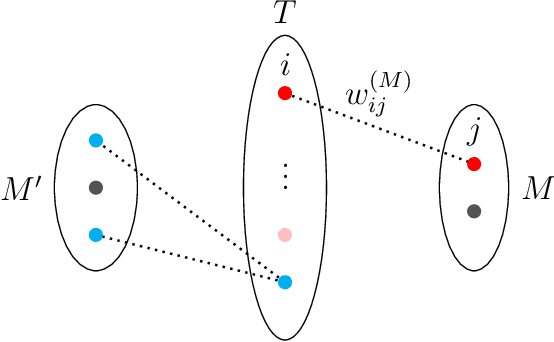
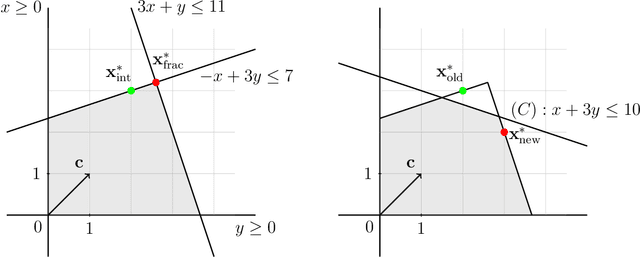

Integer linear programming (ILP) is an elegant approach to solve linear optimization problems, naturally described using integer decision variables. Within the context of physics-inspired machine learning applied to chemistry, we demonstrate the relevance of an ILP formulation to select molecular training sets for predictions of size-extensive properties. We show that our algorithm outperforms existing unsupervised training set selection approaches, especially when predicting properties of molecules larger than those present in the training set. We argue that the reason for the improved performance is due to the selection that is based on the notion of local similarity (i.e., per-atom) and a unique ILP approach that finds optimal solutions efficiently. Altogether, this work provides a practical algorithm to improve the performance of physics-inspired machine learning models and offers insights into the conceptual differences with existing training set selection approaches.
EquiReact: An equivariant neural network for chemical reactions
Dec 13, 2023Equivariant neural networks have considerably improved the accuracy and data-efficiency of predictions of molecular properties. Building on this success, we introduce EquiReact, an equivariant neural network to infer properties of chemical reactions, built from three-dimensional structures of reactants and products. We illustrate its competitive performance on the prediction of activation barriers on the GDB7-22-TS, Cyclo-23-TS and Proparg-21-TS datasets with different regimes according to the inclusion of atom-mapping information. We show that, compared to state-of-the-art models for reaction property prediction, EquiReact offers: (i) a flexible model with reduced sensitivity between atom-mapping regimes, (ii) better extrapolation capabilities to unseen chemistries, (iii) impressive prediction errors for datasets exhibiting subtle variations in three-dimensional geometries of reactants/products, (iv) reduced sensitivity to geometry quality and (iv) excellent data efficiency.