 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDNABERT-2: Efficient Foundation Model and Benchmark For Multi-Species Genome
Jun 26, 2023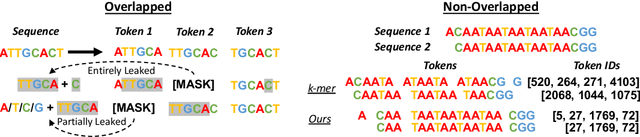
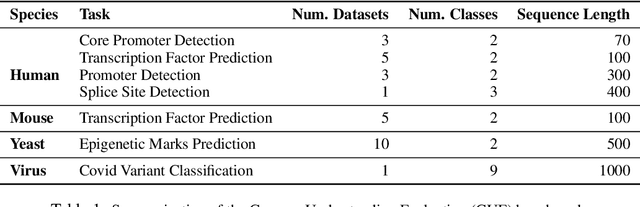

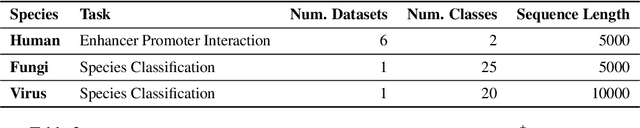
Decoding the linguistic intricacies of the genome is a crucial problem in biology, and pre-trained foundational models such as DNABERT and Nucleotide Transformer have made significant strides in this area. Existing works have largely hinged on k-mer, fixed-length permutations of A, T, C, and G, as the token of the genome language due to its simplicity. However, we argue that the computation and sample inefficiencies introduced by k-mer tokenization are primary obstacles in developing large genome foundational models. We provide conceptual and empirical insights into genome tokenization, building on which we propose to replace k-mer tokenization with Byte Pair Encoding (BPE), a statistics-based data compression algorithm that constructs tokens by iteratively merging the most frequent co-occurring genome segment in the corpus. We demonstrate that BPE not only overcomes the limitations of k-mer tokenization but also benefits from the computational efficiency of non-overlapping tokenization. Based on these insights, we introduce DNABERT-2, a refined genome foundation model that adapts an efficient tokenizer and employs multiple strategies to overcome input length constraints, reduce time and memory expenditure, and enhance model capability. Furthermore, we identify the absence of a comprehensive and standardized benchmark for genome understanding as another significant impediment to fair comparative analysis. In response, we propose the Genome Understanding Evaluation (GUE), a comprehensive multi-species genome classification dataset that amalgamates $28$ distinct datasets across $7$ tasks, with input lengths ranging from $70$ to $1000$. Through comprehensive experiments on the GUE benchmark, we demonstrate that DNABERT-2 achieves comparable performance to the state-of-the-art model with $21 \times$ fewer parameters and approximately $56 \times$ less GPU time in pre-training.