 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePeakNet: Bragg peak finding in X-ray crystallography experiments with U-Net
Mar 24, 2023Serial crystallography at X-ray free electron laser (XFEL) sources has experienced tremendous progress in achieving high data rate in recent times. While this development offers potential to enable novel scientific investigations, such as imaging molecular events at logarithmic timescales, it also poses challenges in regards to real-time data analysis, which involves some degree of data reduction to only save those features or images pertaining to the science on disks. If data reduction is not effective, it could directly result in a substantial increase in facility budgetary requirements, or even hinder the utilization of ultra-high repetition imaging techniques making data analysis unwieldy. Furthermore, an additional challenge involves providing real-time feedback to users derived from real-time data analysis. In the context of serial crystallography, the initial and critical step in real-time data analysis is finding X-ray Bragg peaks from diffraction images. To tackle this challenge, we present PeakNet, a Bragg peak finder that utilizes neural networks and runs about four times faster than Psocake peak finder, while delivering significantly better indexing rates and comparable number of indexed events. We formulated the task of peak finding into a semantic segmentation problem, which is implemented as a classical U-Net architecture. A key advantage of PeakNet is its ability to scale linearly with respect to data volume, making it well-suited for real-time serial crystallography data analysis at high data rates.
Sequence-guided protein structure determination using graph convolutional and recurrent networks
Jul 14, 2020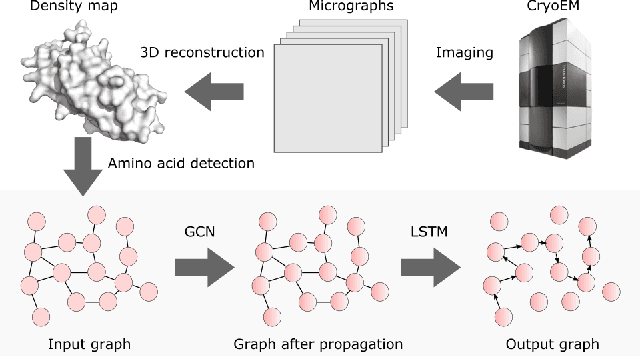
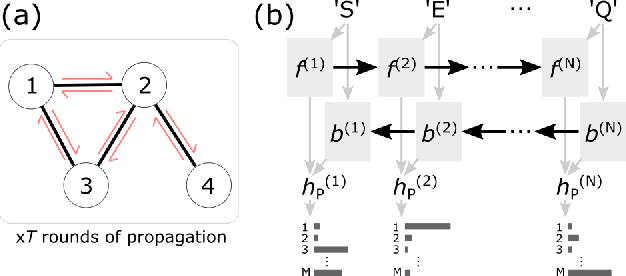
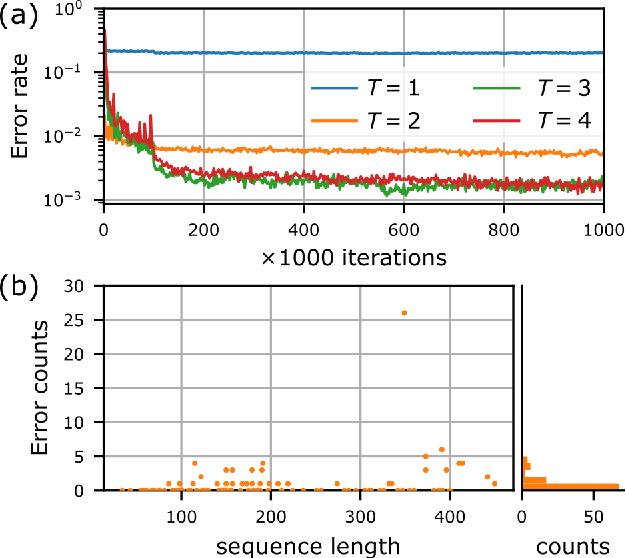
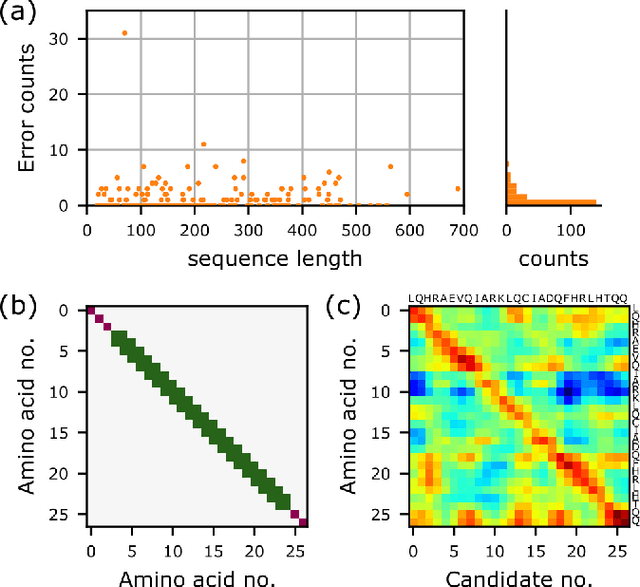
Single particle, cryogenic electron microscopy (cryo-EM) experiments now routinely produce high-resolution data for large proteins and their complexes. Building an atomic model into a cryo-EM density map is challenging, particularly when no structure for the target protein is known a priori. Existing protocols for this type of task often rely on significant human intervention and can take hours to many days to produce an output. Here, we present a fully automated, template-free model building approach that is based entirely on neural networks. We use a graph convolutional network (GCN) to generate an embedding from a set of rotamer-based amino acid identities and candidate 3-dimensional C$\alpha$ locations. Starting from this embedding, we use a bidirectional long short-term memory (LSTM) module to order and label the candidate identities and atomic locations consistent with the input protein sequence to obtain a structural model. Our approach paves the way for determining protein structures from cryo-EM densities at a fraction of the time of existing approaches and without the need for human intervention.
Dual-Reference Design for Holographic Coherent Diffraction Imaging
Feb 07, 2019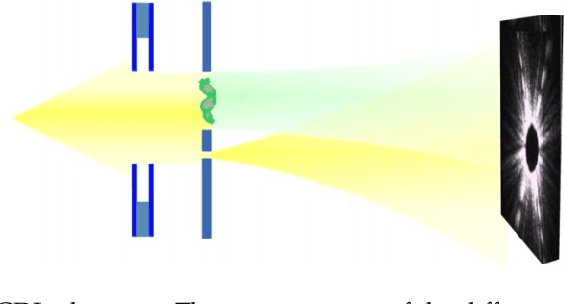
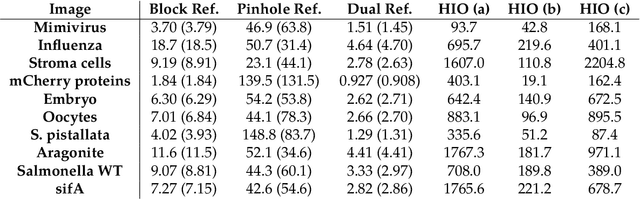
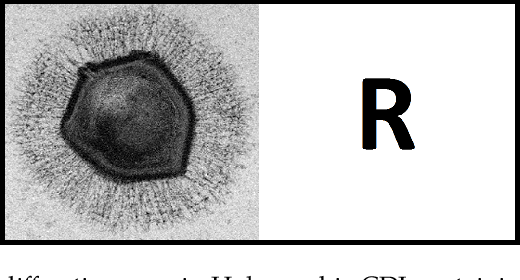

A new reference design is introduced for Holographic Coherent Diffraction Imaging. This consists of two reference portions - being "block" and "pinhole" shaped regions - adjacent to the imaging specimen. Expected error analysis on data following a Poisson shot noise model shows that the dual-reference scheme produces smaller weighting of error across the frequency spectrum than does the leading single-reference schemes. Numerical experiments on simulated data also shows the dual-reference scheme achieving a smaller recovery error than the leading single-reference schemes.