 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFederated Variational Inference for Bayesian Mixture Models
Feb 18, 2025



We present a federated learning approach for Bayesian model-based clustering of large-scale binary and categorical datasets. We introduce a principled 'divide and conquer' inference procedure using variational inference with local merge and delete moves within batches of the data in parallel, followed by 'global' merge moves across batches to find global clustering structures. We show that these merge moves require only summaries of the data in each batch, enabling federated learning across local nodes without requiring the full dataset to be shared. Empirical results on simulated and benchmark datasets demonstrate that our method performs well in comparison to existing clustering algorithms. We validate the practical utility of the method by applying it to large scale electronic health record (EHR) data.
Permutation invariant multi-output Gaussian Processes for drug combination prediction in cancer
Jun 28, 2024
Dose-response prediction in cancer is an active application field in machine learning. Using large libraries of \textit{in-vitro} drug sensitivity screens, the goal is to develop accurate predictive models that can be used to guide experimental design or inform treatment decisions. Building on previous work that makes use of permutation invariant multi-output Gaussian Processes in the context of dose-response prediction for drug combinations, we develop a variational approximation to these models. The variational approximation enables a more scalable model that provides uncertainty quantification and naturally handles missing data. Furthermore, we propose using a deep generative model to encode the chemical space in a continuous manner, enabling prediction for new drugs and new combinations. We demonstrate the performance of our model in a simple setting using a high-throughput dataset and show that the model is able to efficiently borrow information across outputs.
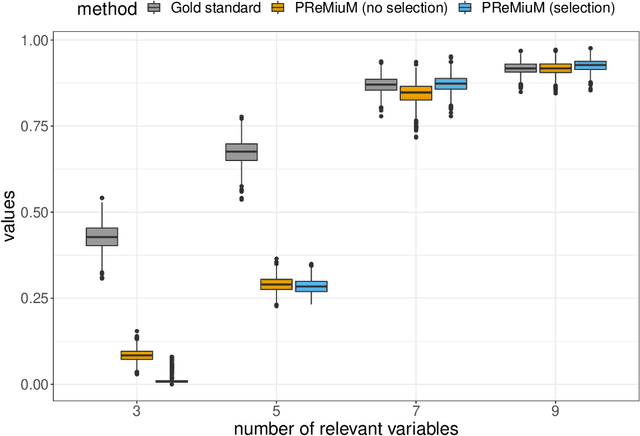
VICatMix: variational Bayesian clustering and variable selection for discrete biomedical data
Jun 23, 2024



Effective clustering of biomedical data is crucial in precision medicine, enabling accurate stratifiction of patients or samples. However, the growth in availability of high-dimensional categorical data, including `omics data, necessitates computationally efficient clustering algorithms. We present VICatMix, a variational Bayesian finite mixture model designed for the clustering of categorical data. The use of variational inference (VI) in its training allows the model to outperform competitors in term of efficiency, while maintaining high accuracy. VICatMix furthermore performs variable selection, enhancing its performance on high-dimensional, noisy data. The proposed model incorporates summarisation and model averaging to mitigate poor local optima in VI, allowing for improved estimation of the true number of clusters simultaneously with feature saliency. We demonstrate the performance of VICatMix with both simulated and real-world data, including applications to datasets from The Cancer Genome Atlas (TCGA), showing its use in cancer subtyping and driver gene discovery. We demonstrate VICatMix's utility in integrative cluster analysis with different `omics datasets, enabling the discovery of novel subtypes. \textbf{Availability:} VICatMix is freely available as an R package, incorporating C++ for faster computation, at \url{https://github.com/j-ackierao/VICatMix}.
Bayesian outcome-guided multi-view mixture models with applications in molecular precision medicine
Mar 01, 2023


Clustering is commonly performed as an initial analysis step for uncovering structure in 'omics datasets, e.g. to discover molecular subtypes of disease. The high-throughput, high-dimensional nature of these datasets means that they provide information on a diverse array of different biomolecular processes and pathways. Different groups of variables (e.g. genes or proteins) will be implicated in different biomolecular processes, and hence undertaking analyses that are limited to identifying just a single clustering partition of the whole dataset is therefore liable to conflate the multiple clustering structures that may arise from these distinct processes. To address this, we propose a multi-view Bayesian mixture model that identifies groups of variables (``views"), each of which defines a distinct clustering structure. We consider applications in stratified medicine, for which our principal goal is to identify clusters of patients that define distinct, clinically actionable disease subtypes. We adopt the semi-supervised, outcome-guided mixture modelling approach of Bayesian profile regression that makes use of a response variable in order to guide inference toward the clusterings that are most relevant in a stratified medicine context. We present the model, together with illustrative simulation examples, and examples from pan-cancer proteomics. We demonstrate how the approach can be used to perform integrative clustering, and consider an example in which different 'omics datasets are integrated in the context of breast cancer subtyping.
Bayesian nonparametric mixture inconsistency for the number of components: How worried should we be in practice?
Jul 29, 2022



We consider the Bayesian mixture of finite mixtures (MFMs) and Dirichlet process mixture (DPM) models for clustering. Recent asymptotic theory has established that DPMs overestimate the number of clusters for large samples and that estimators from both classes of models are inconsistent for the number of clusters under misspecification, but the implications for finite sample analyses are unclear. The final reported estimate after fitting these models is often a single representative clustering obtained using an MCMC summarisation technique, but it is unknown how well such a summary estimates the number of clusters. Here we investigate these practical considerations through simulations and an application to gene expression data, and find that (i) DPMs overestimate the number of clusters even in finite samples, but only to a limited degree that may be correctable using appropriate summaries, and (ii) misspecification can lead to considerable overestimation of the number of clusters in both DPMs and MFMs, but results are nevertheless often still interpretable. We provide recommendations on MCMC summarisation and suggest that although the more appealing asymptotic properties of MFMs provide strong motivation to prefer them, results obtained using MFMs and DPMs are often very similar in practice.
Kernel learning approaches for summarising and combining posterior similarity matrices
Sep 27, 2020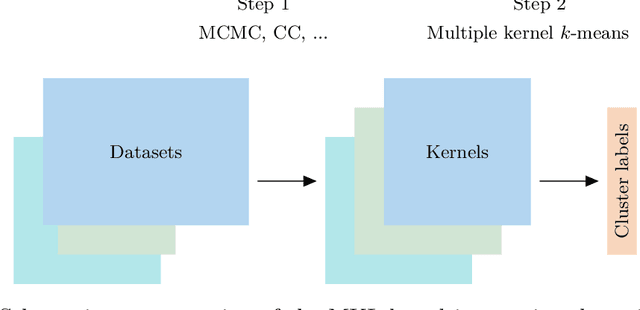

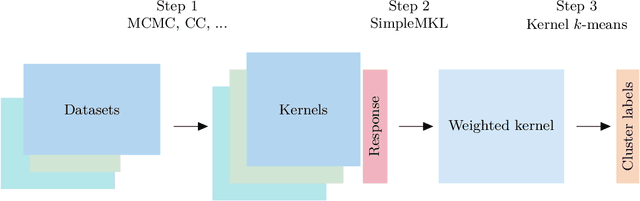

When using Markov chain Monte Carlo (MCMC) algorithms to perform inference for Bayesian clustering models, such as mixture models, the output is typically a sample of clusterings (partitions) drawn from the posterior distribution. In practice, a key challenge is how to summarise this output. Here we build upon the notion of the posterior similarity matrix (PSM) in order to suggest new approaches for summarising the output of MCMC algorithms for Bayesian clustering models. A key contribution of our work is the observation that PSMs are positive semi-definite, and hence can be used to define probabilistically-motivated kernel matrices that capture the clustering structure present in the data. This observation enables us to employ a range of kernel methods to obtain summary clusterings, and otherwise exploit the information summarised by PSMs. For example, if we have multiple PSMs, each corresponding to a different dataset on a common set of statistical units, we may use standard methods for combining kernels in order to perform integrative clustering. We may moreover embed PSMs within predictive kernel models in order to perform outcome-guided data integration. We demonstrate the performances of the proposed methods through a range of simulation studies as well as two real data applications. R code is available at https://github.com/acabassi/combine-psms.
Two-step penalised logistic regression for multi-omic data with an application to cardiometabolic syndrome
Aug 01, 2020



Building classification models that predict a binary class label on the basis of high dimensional multi-omics datasets poses several challenges, due to the typically widely differing characteristics of the data layers in terms of number of predictors, type of data, and levels of noise. Previous research has shown that applying classical logistic regression with elastic-net penalty to these datasets can lead to poor results (Liu et al., 2018). We implement a two-step approach to multi-omic logistic regression in which variable selection is performed on each layer separately and a predictive model is then built using the variables selected in the first step. Here, our approach is compared to other methods that have been developed for the same purpose, and we adapt existing software for multi-omic linear regression (Zhao and Zucknick, 2020) to the logistic regression setting. Extensive simulation studies show that our approach should be preferred if the goal is to select as many relevant predictors as possible, as well as achieving prediction performances comparable to those of the best competitors. Our motivating example is a cardiometabolic syndrome dataset comprising eight 'omic data types for 2 extreme phenotype groups (10 obese and 10 lipodystrophy individuals) and 185 blood donors. Our proposed approach allows us to identify features that characterise cardiometabolic syndrome at the molecular level. R code is available at https://github.com/acabassi/logistic-regression-for-multi-omic-data.
Multiple kernel learning for integrative consensus clustering of genomic datasets
Apr 15, 2019



Diverse applications - particularly in tumour subtyping - have demonstrated the importance of integrative clustering as a means to combine information from multiple high-dimensional omics datasets. Cluster-Of-Clusters Analysis (COCA) is a popular integrative clustering method that has been widely applied in the context of tumour subtyping. However, the properties of COCA have never been systematically explored, and the robustness of this approach to the inclusion of noisy datasets, or datasets that define conflicting clustering structures, is unclear. We rigorously benchmark COCA, and present Kernel Learning Integrative Clustering (KLIC) as an alternative strategy. KLIC frames the challenge of combining clustering structures as a multiple kernel learning problem, in which different datasets each provide a weighted contribution to the final clustering. This allows the contribution of noisy datasets to be down-weighted relative to more informative datasets. We show through extensive simulation studies that KLIC is more robust than COCA in a variety of situations. R code to run KLIC and COCA can be found at https://github.com/acabassi/klic