 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAI-Driven Acoustic Voice Biomarker-Based Hierarchical Classification of Benign Laryngeal Voice Disorders from Sustained Vowels
Dec 31, 2025Benign laryngeal voice disorders affect nearly one in five individuals and often manifest as dysphonia, while also serving as non-invasive indicators of broader physiological dysfunction. We introduce a clinically inspired hierarchical machine learning framework for automated classification of eight benign voice disorders alongside healthy controls, using acoustic features extracted from short, sustained vowel phonations. Experiments utilized 15,132 recordings from 1,261 speakers in the Saarbruecken Voice Database, covering vowels /a/, /i/, and /u/ at neutral, high, low, and gliding pitches. Mirroring clinical triage workflows, the framework operates in three sequential stages: Stage 1 performs binary screening of pathological versus non-pathological voices by integrating convolutional neural network-derived mel-spectrogram features with 21 interpretable acoustic biomarkers; Stage 2 stratifies voices into Healthy, Functional or Psychogenic, and Structural or Inflammatory groups using a cubic support vector machine; Stage 3 achieves fine-grained classification by incorporating probabilistic outputs from prior stages, improving discrimination of structural and inflammatory disorders relative to functional conditions. The proposed system consistently outperformed flat multi-class classifiers and pre-trained self-supervised models, including META HuBERT and Google HeAR, whose generic objectives are not optimized for sustained clinical phonation. By combining deep spectral representations with interpretable acoustic features, the framework enhances transparency and clinical alignment. These results highlight the potential of quantitative voice biomarkers as scalable, non-invasive tools for early screening, diagnostic triage, and longitudinal monitoring of vocal health.
Application of Deep Learning on Single-Cell RNA-sequencing Data Analysis: A Review
Oct 11, 2022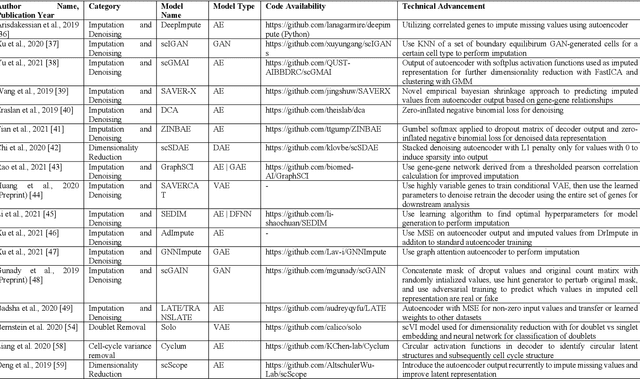

Single-cell RNA-sequencing (scRNA-seq) has become a routinely used technique to quantify the gene expression profile of thousands of single cells simultaneously. Analysis of scRNA-seq data plays an important role in the study of cell states and phenotypes, and has helped elucidate biological processes, such as those occurring during development of complex organisms and improved our understanding of disease states, such as cancer, diabetes, and COVID, among others. Deep learning, a recent advance of artificial intelligence that has been used to address many problems involving large datasets, has also emerged as a promising tool for scRNA-seq data analysis, as it has a capacity to extract informative, compact features from noisy, heterogeneous, and high-dimensional scRNA-seq data to improve downstream analysis. The present review aims at surveying recently developed deep learning techniques in scRNA-seq data analysis, identifying key steps within the scRNA-seq data analysis pipeline that have been advanced by deep learning, and explaining the benefits of deep learning over more conventional analysis tools. Finally, we summarize the challenges in current deep learning approaches faced within scRNA-seq data and discuss potential directions for improvements in deep algorithms for scRNA-seq data analysis.