 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHGTDR: Advancing Drug Repurposing with Heterogeneous Graph Transformers
May 12, 2024Motivation: Drug repurposing is a viable solution for reducing the time and cost associated with drug development. However, thus far, the proposed drug repurposing approaches still need to meet expectations. Therefore, it is crucial to offer a systematic approach for drug repurposing to achieve cost savings and enhance human lives. In recent years, using biological network-based methods for drug repurposing has generated promising results. Nevertheless, these methods have limitations. Primarily, the scope of these methods is generally limited concerning the size and variety of data they can effectively handle. Another issue arises from the treatment of heterogeneous data, which needs to be addressed or converted into homogeneous data, leading to a loss of information. A significant drawback is that most of these approaches lack end-to-end functionality, necessitating manual implementation and expert knowledge in certain stages. Results: We propose a new solution, HGTDR (Heterogeneous Graph Transformer for Drug Repurposing), to address the challenges associated with drug repurposing. HGTDR is a three-step approach for knowledge graph-based drug re-purposing: 1) constructing a heterogeneous knowledge graph, 2) utilizing a heterogeneous graph transformer network, and 3) computing relationship scores using a fully connected network. By leveraging HGTDR, users gain the ability to manipulate input graphs, extract information from diverse entities, and obtain their desired output. In the evaluation step, we demonstrate that HGTDR performs comparably to previous methods. Furthermore, we review medical studies to validate our method's top ten drug repurposing suggestions, which have exhibited promising results. We also demon-strated HGTDR's capability to predict other types of relations through numerical and experimental validation, such as drug-protein and disease-protein inter-relations.
Beyond the Hype: Assessing the Performance, Trustworthiness, and Clinical Suitability of GPT3.5
Jun 28, 2023
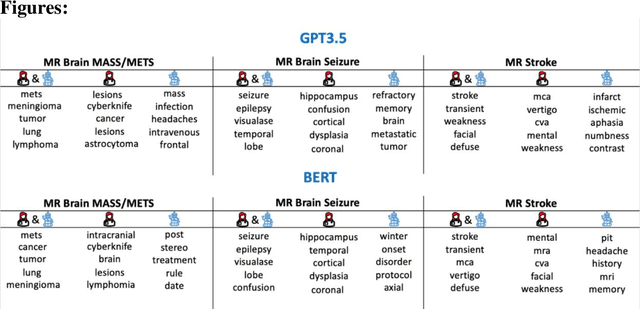
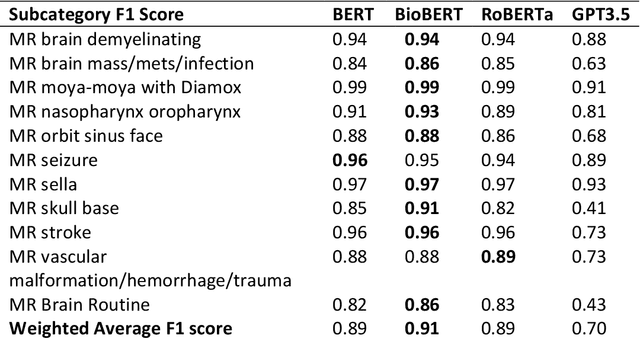
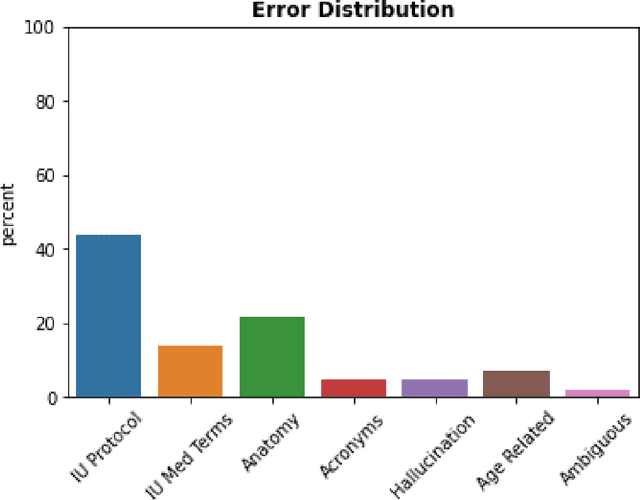
The use of large language models (LLMs) in healthcare is gaining popularity, but their practicality and safety in clinical settings have not been thoroughly assessed. In high-stakes environments like medical settings, trust and safety are critical issues for LLMs. To address these concerns, we present an approach to evaluate the performance and trustworthiness of a GPT3.5 model for medical image protocol assignment. We compare it with a fine-tuned BERT model and a radiologist. In addition, we have a radiologist review the GPT3.5 output to evaluate its decision-making process. Our evaluation dataset consists of 4,700 physician entries across 11 imaging protocol classes spanning the entire head. Our findings suggest that the GPT3.5 performance falls behind BERT and a radiologist. However, GPT3.5 outperforms BERT in its ability to explain its decision, detect relevant word indicators, and model calibration. Furthermore, by analyzing the explanations of GPT3.5 for misclassifications, we reveal systematic errors that need to be resolved to enhance its safety and suitability for clinical use.
UniSent: Universal Adaptable Sentiment Lexica for 1000+ Languages
Apr 21, 2019
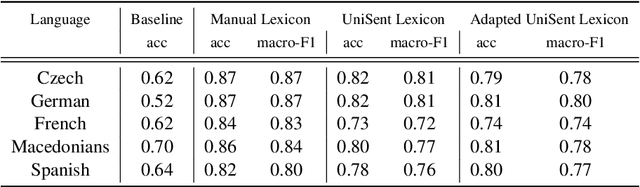
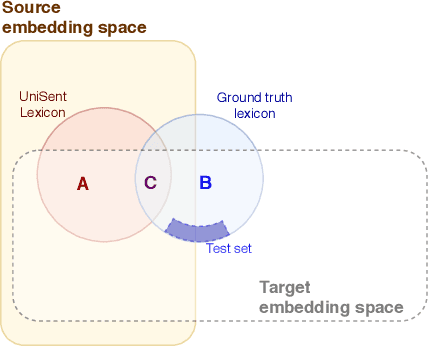
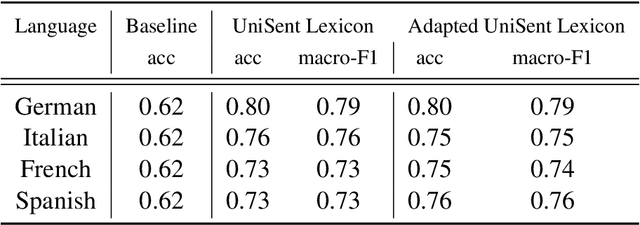
In this paper, we introduce UniSent a universal sentiment lexica for 1000 languages created using an English sentiment lexicon and a massively parallel corpus in the Bible domain. To the best of our knowledge, UniSent is the largest sentiment resource to date in terms of number of covered languages, including many low resource languages. To create UniSent, we propose Adapted Sentiment Pivot, a novel method that combines annotation projection, vocabulary expansion, and unsupervised domain adaptation. We evaluate the quality of UniSent for Macedonian, Czech, German, Spanish, and French and show that its quality is comparable to manually or semi-manually created sentiment resources. With the publication of this paper, we release UniSent lexica as well as Adapted Sentiment Pivot related codes. method.
ProtVec: A Continuous Distributed Representation of Biological Sequences
May 26, 2016



We introduce a new representation and feature extraction method for biological sequences. Named bio-vectors (BioVec) to refer to biological sequences in general with protein-vectors (ProtVec) for proteins (amino-acid sequences) and gene-vectors (GeneVec) for gene sequences, this representation can be widely used in applications of deep learning in proteomics and genomics. In the present paper, we focus on protein-vectors that can be utilized in a wide array of bioinformatics investigations such as family classification, protein visualization, structure prediction, disordered protein identification, and protein-protein interaction prediction. In this method, we adopt artificial neural network approaches and represent a protein sequence with a single dense n-dimensional vector. To evaluate this method, we apply it in classification of 324,018 protein sequences obtained from Swiss-Prot belonging to 7,027 protein families, where an average family classification accuracy of 93%+-0.06% is obtained, outperforming existing family classification methods. In addition, we use ProtVec representation to predict disordered proteins from structured proteins. Two databases of disordered sequences are used: the DisProt database as well as a database featuring the disordered regions of nucleoporins rich with phenylalanine-glycine repeats (FG-Nups). Using support vector machine classifiers, FG-Nup sequences are distinguished from structured protein sequences found in Protein Data Bank (PDB) with a 99.8% accuracy, and unstructured DisProt sequences are differentiated from structured DisProt sequences with 100.0% accuracy. These results indicate that by only providing sequence data for various proteins into this model, accurate information about protein structure can be determined.
Comparing Fifty Natural Languages and Twelve Genetic Languages Using Word Embedding Language Divergence (WELD) as a Quantitative Measure of Language Distance
Apr 28, 2016
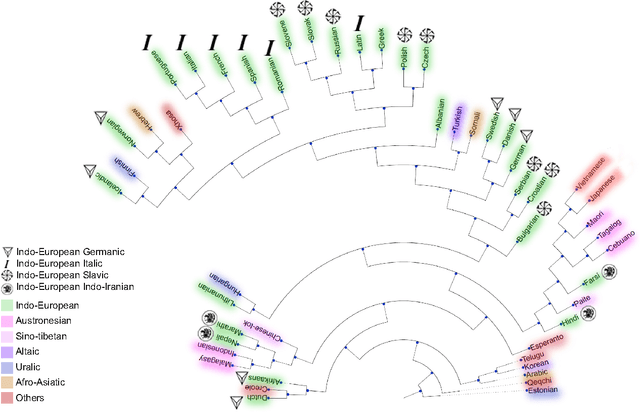

We introduce a new measure of distance between languages based on word embedding, called word embedding language divergence (WELD). WELD is defined as divergence between unified similarity distribution of words between languages. Using such a measure, we perform language comparison for fifty natural languages and twelve genetic languages. Our natural language dataset is a collection of sentence-aligned parallel corpora from bible translations for fifty languages spanning a variety of language families. Although we use parallel corpora, which guarantees having the same content in all languages, interestingly in many cases languages within the same family cluster together. In addition to natural languages, we perform language comparison for the coding regions in the genomes of 12 different organisms (4 plants, 6 animals, and two human subjects). Our result confirms a significant high-level difference in the genetic language model of humans/animals versus plants. The proposed method is a step toward defining a quantitative measure of similarity between languages, with applications in languages classification, genre identification, dialect identification, and evaluation of translations.