 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Histopathology Study Comparing Contrastive Semi-Supervised and Fully Supervised Learning
Nov 10, 2021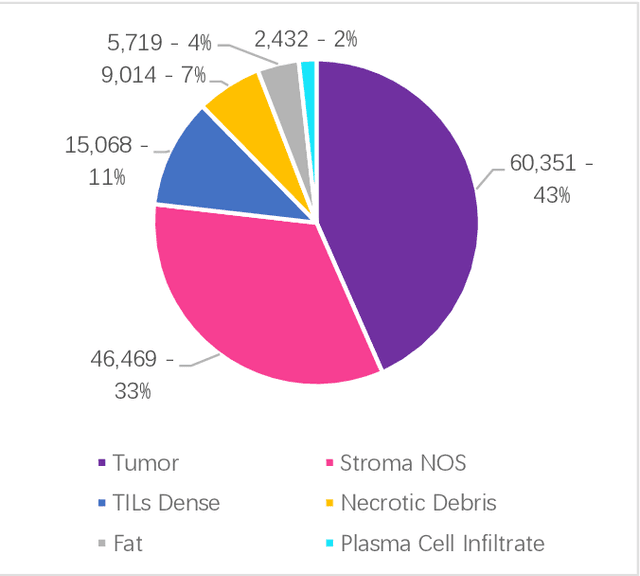
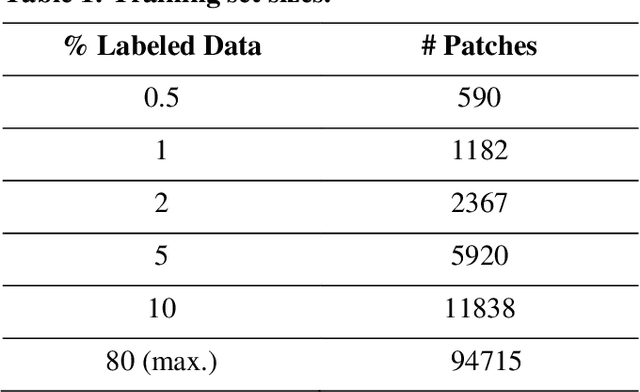
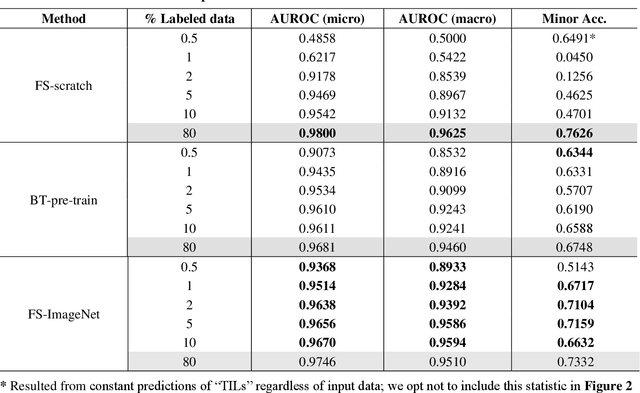
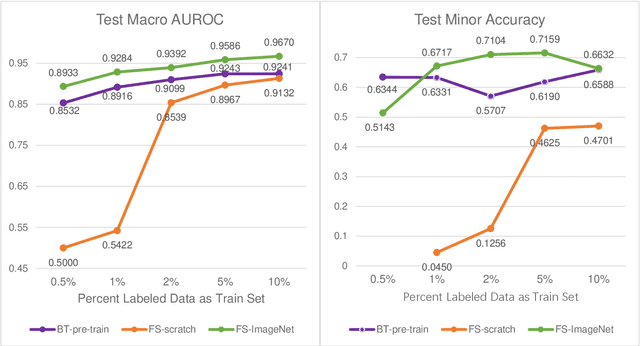
Data labeling is often the most challenging task when developing computational pathology models. Pathologist participation is necessary to generate accurate labels, and the limitations on pathologist time and demand for large, labeled datasets has led to research in areas including weakly supervised learning using patient-level labels, machine assisted annotation and active learning. In this paper we explore self-supervised learning to reduce labeling burdens in computational pathology. We explore this in the context of classification of breast cancer tissue using the Barlow Twins approach, and we compare self-supervision with alternatives like pre-trained networks in low-data scenarios. For the task explored in this paper, we find that ImageNet pre-trained networks largely outperform the self-supervised representations obtained using Barlow Twins.
NuCLS: A scalable crowdsourcing, deep learning approach and dataset for nucleus classification, localization and segmentation
Feb 18, 2021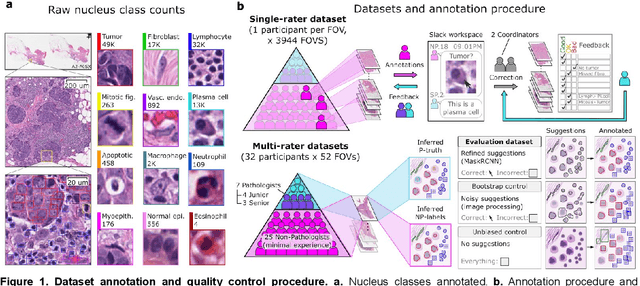
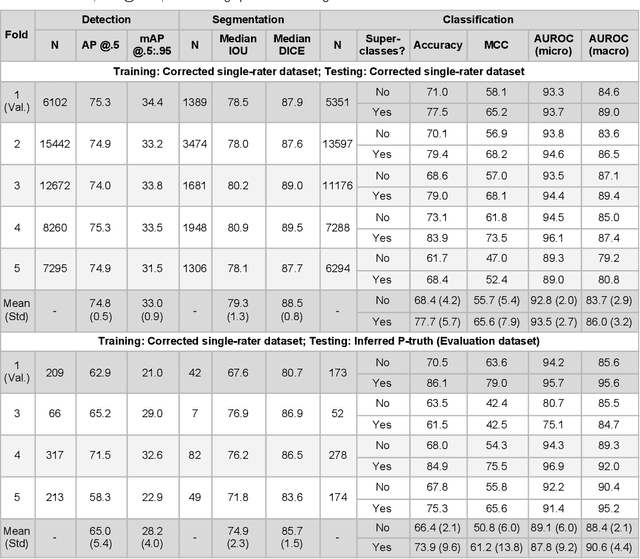
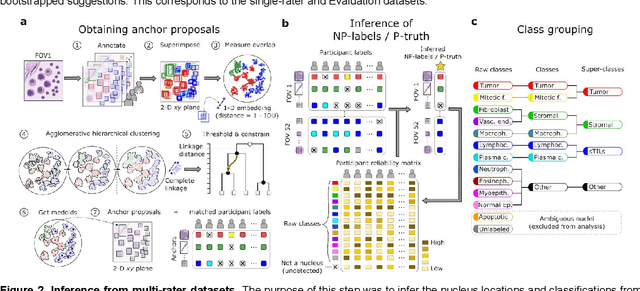
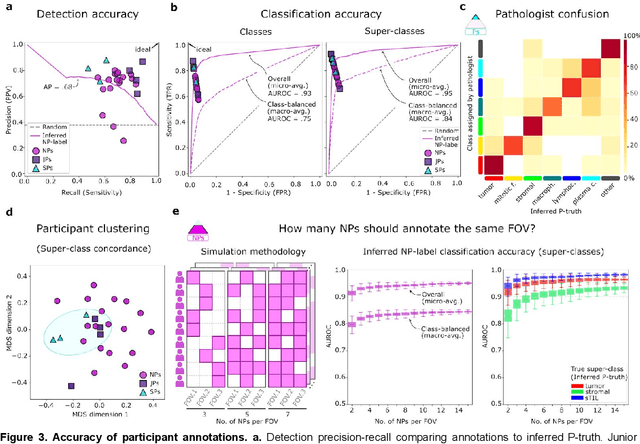
High-resolution mapping of cells and tissue structures provides a foundation for developing interpretable machine-learning models for computational pathology. Deep learning algorithms can provide accurate mappings given large numbers of labeled instances for training and validation. Generating adequate volume of quality labels has emerged as a critical barrier in computational pathology given the time and effort required from pathologists. In this paper we describe an approach for engaging crowds of medical students and pathologists that was used to produce a dataset of over 220,000 annotations of cell nuclei in breast cancers. We show how suggested annotations generated by a weak algorithm can improve the accuracy of annotations generated by non-experts and can yield useful data for training segmentation algorithms without laborious manual tracing. We systematically examine interrater agreement and describe modifications to the MaskRCNN model to improve cell mapping. We also describe a technique we call Decision Tree Approximation of Learned Embeddings (DTALE) that leverages nucleus segmentations and morphologic features to improve the transparency of nucleus classification models. The annotation data produced in this study are freely available for algorithm development and benchmarking at: https://sites.google.com/view/nucls.
HistomicsML2.0: Fast interactive machine learning for whole slide imaging data
Jan 30, 2020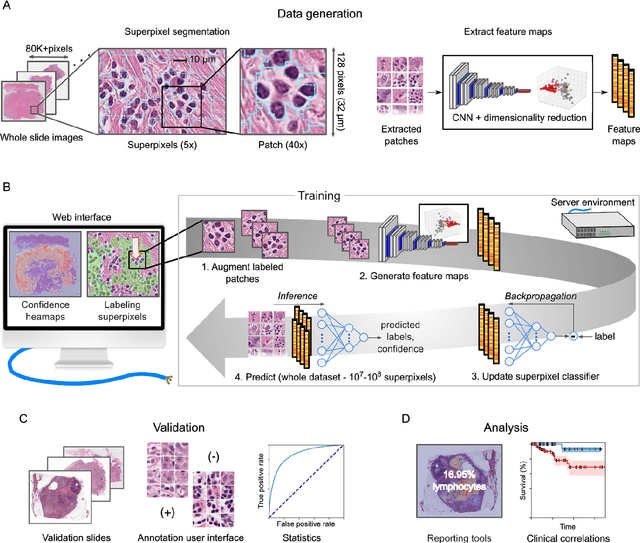
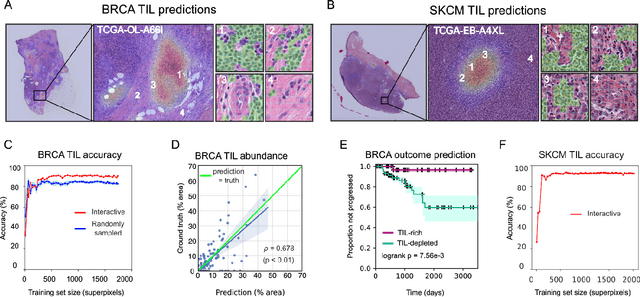
Extracting quantitative phenotypic information from whole-slide images presents significant challenges for investigators who are not experienced in developing image analysis algorithms. We present new software that enables rapid learn-by-example training of machine learning classifiers for detection of histologic patterns in whole-slide imaging datasets. HistomicsML2.0 uses convolutional networks to be readily adaptable to a variety of applications, provides a web-based user interface, and is available as a software container to simplify deployment.