 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMICCAI-CDMRI 2023 QuantConn Challenge Findings on Achieving Robust Quantitative Connectivity through Harmonized Preprocessing of Diffusion MRI
Nov 14, 2024



White matter alterations are increasingly implicated in neurological diseases and their progression. International-scale studies use diffusion-weighted magnetic resonance imaging (DW-MRI) to qualitatively identify changes in white matter microstructure and connectivity. Yet, quantitative analysis of DW-MRI data is hindered by inconsistencies stemming from varying acquisition protocols. There is a pressing need to harmonize the preprocessing of DW-MRI datasets to ensure the derivation of robust quantitative diffusion metrics across acquisitions. In the MICCAI-CDMRI 2023 QuantConn challenge, participants were provided raw data from the same individuals collected on the same scanner but with two different acquisitions and tasked with preprocessing the DW-MRI to minimize acquisition differences while retaining biological variation. Submissions are evaluated on the reproducibility and comparability of cross-acquisition bundle-wise microstructure measures, bundle shape features, and connectomics. The key innovations of the QuantConn challenge are that (1) we assess bundles and tractography in the context of harmonization for the first time, (2) we assess connectomics in the context of harmonization for the first time, and (3) we have 10x additional subjects over prior harmonization challenge, MUSHAC and 100x over SuperMUDI. We find that bundle surface area, fractional anisotropy, connectome assortativity, betweenness centrality, edge count, modularity, nodal strength, and participation coefficient measures are most biased by acquisition and that machine learning voxel-wise correction, RISH mapping, and NeSH methods effectively reduce these biases. In addition, microstructure measures AD, MD, RD, bundle length, connectome density, efficiency, and path length are least biased by these acquisition differences.
* Accepted for publication at the Journal of Machine Learning for Biomedical Imaging (MELBA) https://melba-journal.org/2024/019
Multi-dimensional Parameter Space Exploration for Streamline-specific Tractography
Aug 09, 2024One of the unspoken challenges of tractography is choosing the right parameters for a given dataset or bundle. In order to tackle this challenge, we explore the multi-dimensional parameter space of tractography using streamline-specific parameters (SSP). We 1) validate a state-of-the-art probabilistic tracking method using per-streamline parameters on synthetic data, and 2) show how we can gain insights into the parameter space by focusing on streamline acceptance using real-world data. We demonstrate the potential added value of SSP to the current state of tractography by showing how SSP can be used to reveal patterns in the parameter space.
Neural Spherical Harmonics for structurally coherent continuous representation of diffusion MRI signal
Aug 23, 2023We present a novel way to model diffusion magnetic resonance imaging (dMRI) datasets, that benefits from the structural coherence of the human brain while only using data from a single subject. Current methods model the dMRI signal in individual voxels, disregarding the intervoxel coherence that is present. We use a neural network to parameterize a spherical harmonics series (NeSH) to represent the dMRI signal of a single subject from the Human Connectome Project dataset, continuous in both the angular and spatial domain. The reconstructed dMRI signal using this method shows a more structurally coherent representation of the data. Noise in gradient images is removed and the fiber orientation distribution functions show a smooth change in direction along a fiber tract. We showcase how the reconstruction can be used to calculate mean diffusivity, fractional anisotropy, and total apparent fiber density. These results can be achieved with a single model architecture, tuning only one hyperparameter. In this paper we also demonstrate how upsampling in both the angular and spatial domain yields reconstructions that are on par or better than existing methods.
Tractometry-based Anomaly Detection for Single-subject White Matter Analysis
May 25, 2020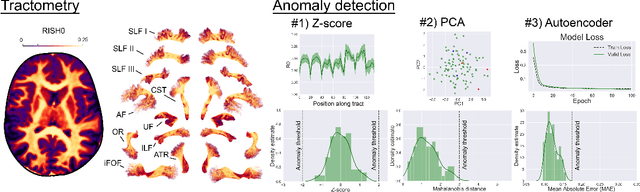
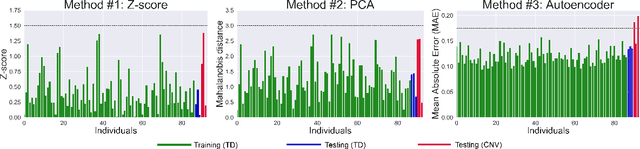
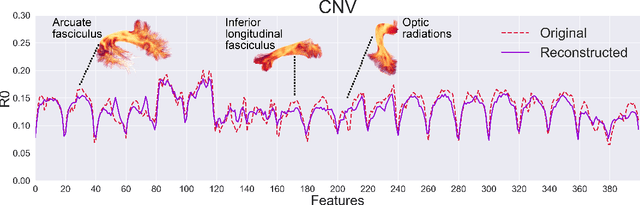
There is an urgent need for a paradigm shift from group-wise comparisons to individual diagnosis in diffusion MRI (dMRI) to enable the analysis of rare cases and clinically-heterogeneous groups. Deep autoencoders have shown great potential to detect anomalies in neuroimaging data. We present a framework that operates on the manifold of white matter (WM) pathways to learn normative microstructural features, and discriminate those at genetic risk from controls in a paediatric population.
* Medical Imaging with Deep Learning (MIDL2020) Conference Short Paper