 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHierarchical Pooling and Explainability in Graph Neural Networks for Tumor and Tissue-of-Origin Classification Using RNA-seq Data
Jan 10, 2026This study explores the use of graph neural networks (GNNs) with hierarchical pooling and multiple convolution layers for cancer classification based on RNA-seq data. We combine gene expression data from The Cancer Genome Atlas (TCGA) with a precomputed STRING protein-protein interaction network to classify tissue origin and distinguish between normal and tumor samples. The model employs Chebyshev graph convolutions (K=2) and weighted pooling layers, aggregating gene clusters into 'supernodes' across multiple coarsening levels. This approach enables dimensionality reduction while preserving meaningful interactions. Saliency methods were applied to interpret the model by identifying key genes and biological processes relevant to cancer. Our findings reveal that increasing the number of convolution and pooling layers did not enhance classification performance. The highest F1-macro score (0.978) was achieved with a single pooling layer. However, adding more layers resulted in over-smoothing and performance degradation. However, the model proved highly interpretable through gradient methods, identifying known cancer-related genes and highlighting enriched biological processes, and its hierarchical structure can be used to develop new explainable architectures. Overall, while deeper GNN architectures did not improve performance, the hierarchical pooling structure provided valuable insights into tumor biology, making GNNs a promising tool for cancer biomarker discovery and interpretation
Comparing Cluster-Based Cross-Validation Strategies for Machine Learning Model Evaluation
Jul 30, 2025



Cross-validation plays a fundamental role in Machine Learning, enabling robust evaluation of model performance and preventing overestimation on training and validation data. However, one of its drawbacks is the potential to create data subsets (folds) that do not adequately represent the diversity of the original dataset, which can lead to biased performance estimates. The objective of this work is to deepen the investigation of cluster-based cross-validation strategies by analyzing the performance of different clustering algorithms through experimental comparison. Additionally, a new cross-validation technique that combines Mini Batch K-Means with class stratification is proposed. Experiments were conducted on 20 datasets (both balanced and imbalanced) using four supervised learning algorithms, comparing cross-validation strategies in terms of bias, variance, and computational cost. The technique that uses Mini Batch K-Means with class stratification outperformed others in terms of bias and variance on balanced datasets, though it did not significantly reduce computational cost. On imbalanced datasets, traditional stratified cross-validation consistently performed better, showing lower bias, variance, and computational cost, making it a safe choice for performance evaluation in scenarios with class imbalance. In the comparison of different clustering algorithms, no single algorithm consistently stood out as superior. Overall, this work contributes to improving predictive model evaluation strategies by providing a deeper understanding of the potential of cluster-based data splitting techniques and reaffirming the effectiveness of well-established strategies like stratified cross-validation. Moreover, it highlights perspectives for increasing the robustness and reliability of model evaluations, especially in datasets with clustering characteristics.
Exploring the Feasibility of AI-Assisted Spine MRI Protocol Optimization Using DICOM Image Metadata
Feb 04, 2025



Artificial intelligence (AI) is increasingly being utilized to optimize magnetic resonance imaging (MRI) protocols. Given that image details are critical for diagnostic accuracy, optimizing MRI acquisition protocols is essential for enhancing image quality. While medical physicists are responsible for this optimization, the variability in equipment usage and the wide range of MRI protocols in clinical settings pose significant challenges. This study aims to validate the application of AI in optimizing MRI protocols using dynamic data from clinical practice, specifically DICOM metadata. To achieve this, four MRI spine exam databases were created, with the target attribute being the binary classification of image quality (good or bad). Five AI models were trained to identify trends in acquisition parameters that influence image quality, grounded in MRI theory. These trends were analyzed using SHAP graphs. The models achieved F1 performance ranging from 77% to 93% for datasets containing 292 or more instances, with the observed trends aligning with MRI theory. The models effectively reflected the practical realities of clinical MRI settings, offering a valuable tool for medical physicists in quality control tasks. In conclusion, AI has demonstrated its potential to optimize MRI protocols, supporting medical physicists in improving image quality and enhancing the efficiency of quality control in clinical practice.
Graph Neural Networks for Heart Failure Prediction on an EHR-Based Patient Similarity Graph
Nov 29, 2024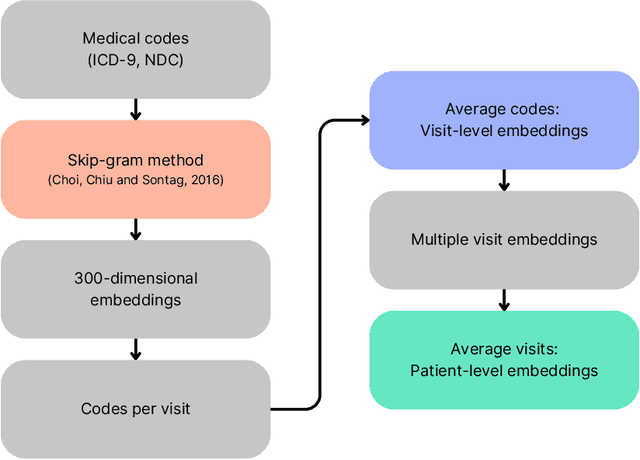
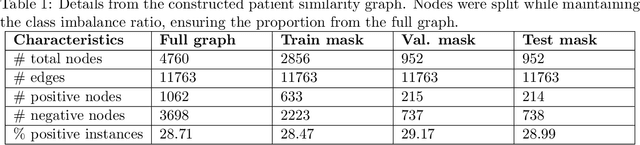

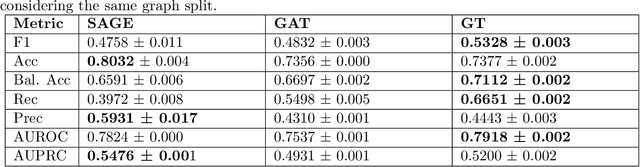
Objective: In modern healthcare, accurately predicting diseases is a crucial matter. This study introduces a novel approach using graph neural networks (GNNs) and a Graph Transformer (GT) to predict the incidence of heart failure (HF) on a patient similarity graph at the next hospital visit. Materials and Methods: We used electronic health records (EHR) from the MIMIC-III dataset and applied the K-Nearest Neighbors (KNN) algorithm to create a patient similarity graph using embeddings from diagnoses, procedures, and medications. Three models - GraphSAGE, Graph Attention Network (GAT), and Graph Transformer (GT) - were implemented to predict HF incidence. Model performance was evaluated using F1 score, AUROC, and AUPRC metrics, and results were compared against baseline algorithms. An interpretability analysis was performed to understand the model's decision-making process. Results: The GT model demonstrated the best performance (F1 score: 0.5361, AUROC: 0.7925, AUPRC: 0.5168). Although the Random Forest (RF) baseline achieved a similar AUPRC value, the GT model offered enhanced interpretability due to the use of patient relationships in the graph structure. A joint analysis of attention weights, graph connectivity, and clinical features provided insight into model predictions across different classification groups. Discussion and Conclusion: Graph-based approaches such as GNNs provide an effective framework for predicting HF. By leveraging a patient similarity graph, GNNs can capture complex relationships in EHR data, potentially improving prediction accuracy and clinical interpretability.
Acoustic Identification of Ae. aegypti Mosquitoes using Smartphone Apps and Residual Convolutional Neural Networks
Jun 16, 2023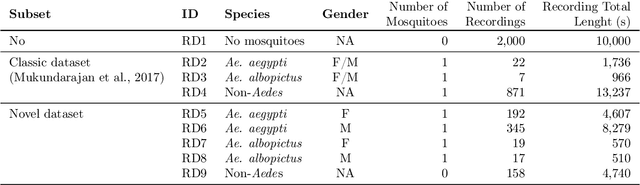
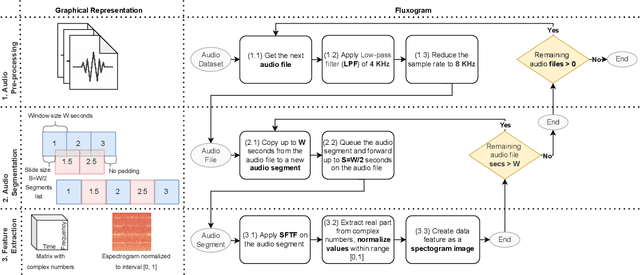

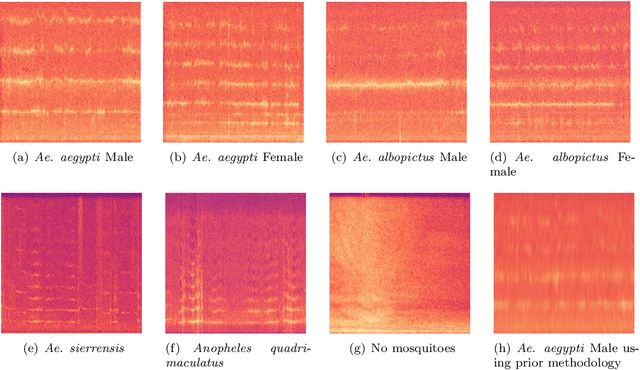
In this paper, we advocate in favor of smartphone apps as low-cost, easy-to-deploy solution for raising awareness among the population on the proliferation of Aedes aegypti mosquitoes. Nevertheless, devising such a smartphone app is challenging, for many reasons, including the required maturity level of techniques for identifying mosquitoes based on features that can be captured using smartphone resources. In this paper, we identify a set of (non-exhaustive) requirements that smartphone apps must meet to become an effective tooling in the fight against Ae. aegypti, and advance the state-of-the-art with (i) a residual convolutional neural network for classifying Ae. aegypti mosquitoes from their wingbeat sound, (ii) a methodology for reducing the influence of background noise in the classification process, and (iii) a dataset for benchmarking solutions for detecting Ae. aegypti mosquitoes from wingbeat sound recordings. From the analysis of accuracy and recall, we provide evidence that convolutional neural networks have potential as a cornerstone for tracking mosquito apps for smartphones.
Machine learning methods for prediction of cancer driver genes: a survey paper
Sep 28, 2021
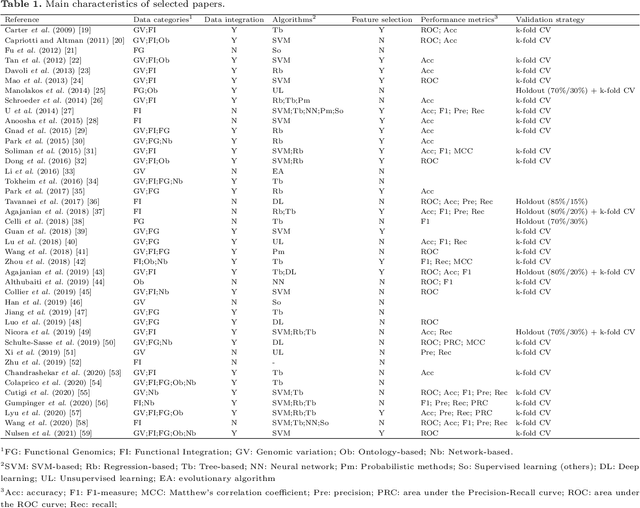

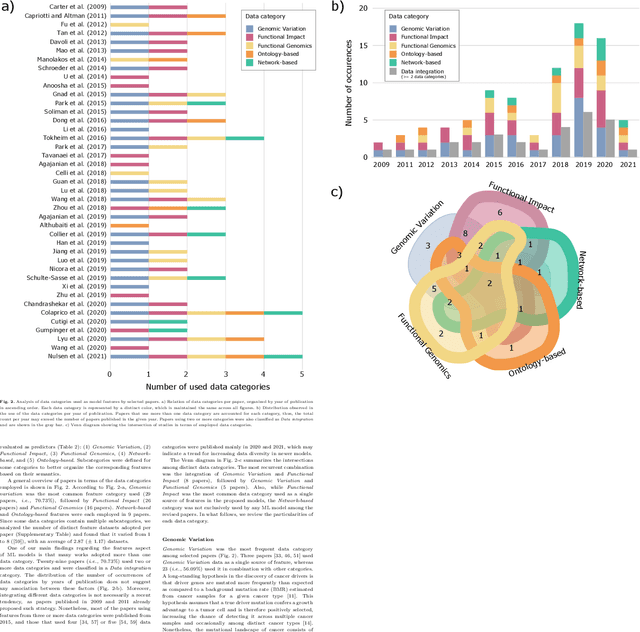
Identifying the genes and mutations that drive the emergence of tumors is a major step to improve understanding of cancer and identify new directions for disease diagnosis and treatment. Despite the large volume of genomics data, the precise detection of driver mutations and their carrying genes, known as cancer driver genes, from the millions of possible somatic mutations remains a challenge. Computational methods play an increasingly important role in identifying genomic patterns associated with cancer drivers and developing models to predict driver events. Machine learning (ML) has been the engine behind many of these efforts and provides excellent opportunities for tackling remaining gaps in the field. Thus, this survey aims to perform a comprehensive analysis of ML-based computational approaches to identify cancer driver mutations and genes, providing an integrated, panoramic view of the broad data and algorithmic landscape within this scientific problem. We discuss how the interactions among data types and ML algorithms have been explored in previous solutions and outline current analytical limitations that deserve further attention from the scientific community. We hope that by helping readers become more familiar with significant developments in the field brought by ML, we may inspire new researchers to address open problems and advance our knowledge towards cancer driver discovery.
A Hybrid Ensemble Feature Selection Design for Candidate Biomarkers Discovery from Transcriptome Profiles
Jul 31, 2021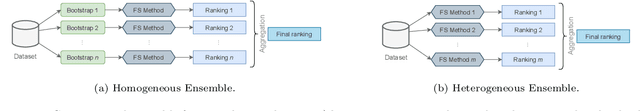

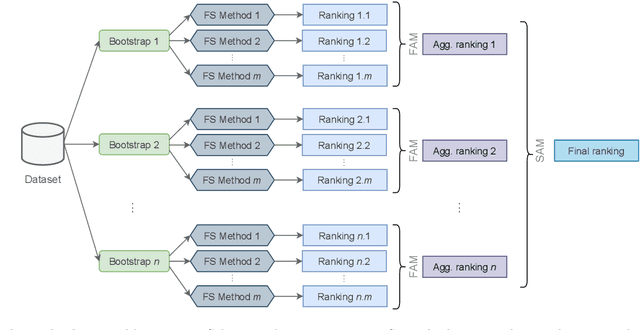
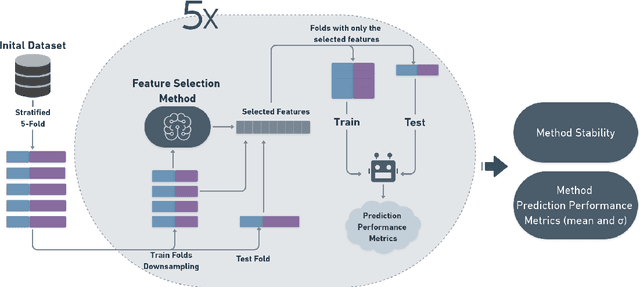
The discovery of disease biomarkers from gene expression data has been greatly advanced by feature selection (FS) methods, especially using ensemble FS (EFS) strategies with perturbation at the data level (i.e., homogeneous, Hom-EFS) or method level (i.e., heterogeneous, Het-EFS). Here we proposed a Hybrid EFS (Hyb-EFS) design that explores both types of perturbation to improve the stability and the predictive power of candidate biomarkers. With this, Hyb-EFS aims to disrupt associations of good performance with a single dataset, single algorithm, or a specific combination of both, which is particularly interesting for better reproducibility of genomic biomarkers. We investigated the adequacy of our approach for microarray data related to four types of cancer, carrying out an extensive comparison with other ensemble and single FS approaches. Five FS methods were used in our experiments: Wx, Symmetrical Uncertainty (SU), Gain Ratio (GR), Characteristic Direction (GeoDE), and ReliefF. We observed that the Hyb-EFS and Het-EFS approaches attenuated the large performance variation observed for most single FS and Hom-EFS across distinct datasets. Also, the Hyb-EFS improved upon the stability of the Het-EFS within our domain. Comparing the Hyb-EFS and Het-EFS composed of the top-performing selectors (Wx, GR, and SU), our hybrid approach surpassed the equivalent heterogeneous design and the best Hom-EFS (Hom-Wx). Interestingly, the rankings produced by our Hyb-EFS reached greater biological plausibility, with a notably high enrichment for cancer-related genes and pathways. Thus, our experiments suggest the potential of the proposed Hybrid EFS design in discovering candidate biomarkers from microarray data. Finally, we provide an open-source framework to support similar analyses in other domains, both as a user-friendly application and a plain Python package.
Detecting Aedes Aegypti Mosquitoes through Audio Classification with Convolutional Neural Networks
Aug 19, 2020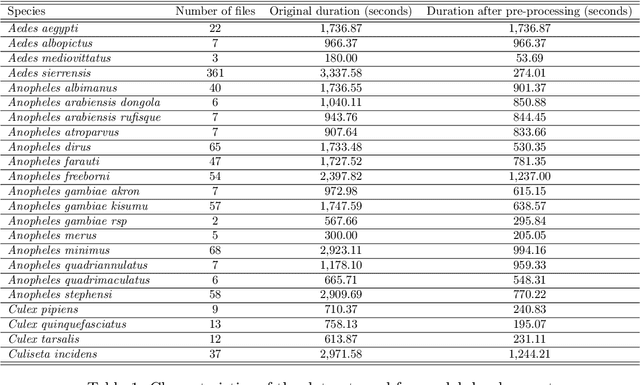
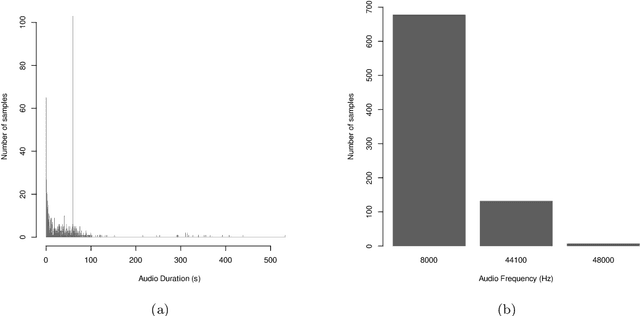
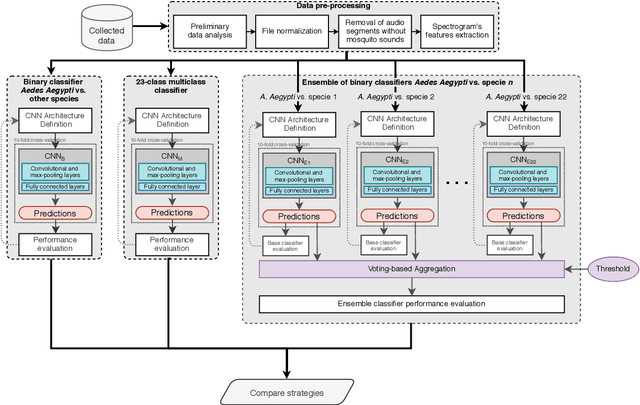
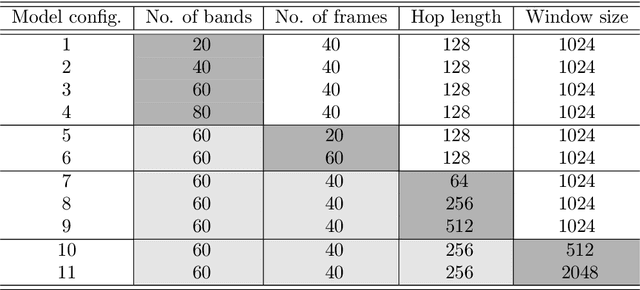
The incidence of mosquito-borne diseases is significant in under-developed regions, mostly due to the lack of resources to implement aggressive control measurements against mosquito proliferation. A potential strategy to raise community awareness regarding mosquito proliferation is building a live map of mosquito incidences using smartphone apps and crowdsourcing. In this paper, we explore the possibility of identifying Aedes aegypti mosquitoes using machine learning techniques and audio analysis captured from commercially available smartphones. In summary, we downsampled Aedes aegypti wingbeat recordings and used them to train a convolutional neural network (CNN) through supervised learning. As a feature, we used the recording spectrogram to represent the mosquito wingbeat frequency over time visually. We trained and compared three classifiers: a binary, a multiclass, and an ensemble of binary classifiers. In our evaluation, the binary and ensemble models achieved accuracy of 97.65% ($\pm$ 0.55) and 94.56% ($\pm$ 0.77), respectively, whereas the multiclass had an accuracy of 78.12% ($\pm$ 2.09). The best sensitivity was observed in the ensemble approach (96.82% $\pm$ 1.62), followed by the multiclass for the particular case of Aedes aegypti (90.23% $\pm$ 3.83) and the binary (88.49% $\pm$ 6.68). The binary classifier and the multiclass classifier presented the best balance between precision and recall, with F1-measure close to 90%. Although the ensemble classifier achieved the lowest precision, thus impairing its F1-measure (79.95% $\pm$ 2.13), it was the most powerful classifier to detect Aedes aegypti in our dataset.
EPGAT: Gene Essentiality Prediction With Graph Attention Networks
Jul 19, 2020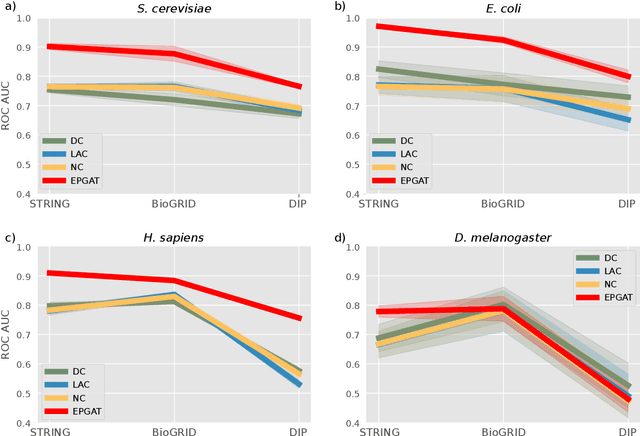
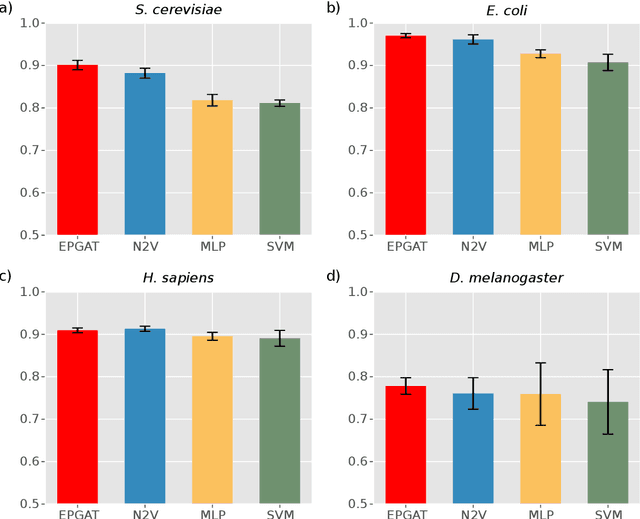
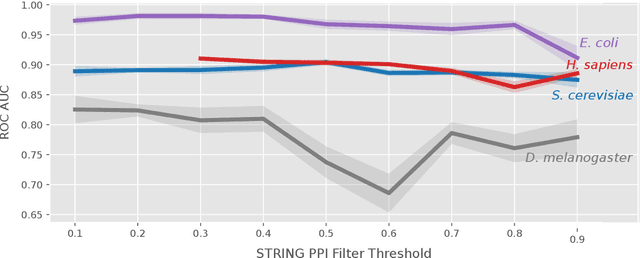
The identification of essential genes/proteins is a critical step towards a better understanding of human biology and pathology. Computational approaches helped to mitigate experimental constraints by exploring machine learning (ML) methods and the correlation of essentiality with biological information, especially protein-protein interaction (PPI) networks, to predict essential genes. Nonetheless, their performance is still limited, as network-based centralities are not exclusive proxies of essentiality, and traditional ML methods are unable to learn from non-Euclidean domains such as graphs. Given these limitations, we proposed EPGAT, an approach for essentiality prediction based on Graph Attention Networks (GATs), which are attention-based Graph Neural Networks (GNNs) that operate on graph-structured data. Our model directly learns patterns of gene essentiality from PPI networks, integrating additional evidence from multiomics data encoded as node attributes. We benchmarked EPGAT for four organisms, including humans, accurately predicting gene essentiality with AUC score ranging from 0.78 to 0.97. Our model significantly outperformed network-based and shallow ML-based methods and achieved a very competitive performance against the state-of-the-art node2vec embedding method. Notably, EPGAT was the most robust approach in scenarios with limited and imbalanced training data. Thus, the proposed approach offers a powerful and effective way to identify essential genes and proteins.