 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA survey of probabilistic generative frameworks for molecular simulations
Nov 14, 2024



Generative artificial intelligence is now a widely used tool in molecular science. Despite the popularity of probabilistic generative models, numerical experiments benchmarking their performance on molecular data are lacking. In this work, we introduce and explain several classes of generative models, broadly sorted into two categories: flow-based models and diffusion models. We select three representative models: Neural Spline Flows, Conditional Flow Matching, and Denoising Diffusion Probabilistic Models, and examine their accuracy, computational cost, and generation speed across datasets with tunable dimensionality, complexity, and modal asymmetry. Our findings are varied, with no one framework being the best for all purposes. In a nutshell, (i) Neural Spline Flows do best at capturing mode asymmetry present in low-dimensional data, (ii) Conditional Flow Matching outperforms other models for high-dimensional data with low complexity, and (iii) Denoising Diffusion Probabilistic Models appears the best for low-dimensional data with high complexity. Our datasets include a Gaussian mixture model and the dihedral torsion angle distribution of the Aib\textsubscript{9} peptide, generated via a molecular dynamics simulation. We hope our taxonomy of probabilistic generative frameworks and numerical results may guide model selection for a wide range of molecular tasks.
Generative artificial intelligence for computational chemistry: a roadmap to predicting emergent phenomena
Sep 04, 2024The recent surge in Generative Artificial Intelligence (AI) has introduced exciting possibilities for computational chemistry. Generative AI methods have made significant progress in sampling molecular structures across chemical species, developing force fields, and speeding up simulations. This Perspective offers a structured overview, beginning with the fundamental theoretical concepts in both Generative AI and computational chemistry. It then covers widely used Generative AI methods, including autoencoders, generative adversarial networks, reinforcement learning, flow models and language models, and highlights their selected applications in diverse areas including force field development, and protein/RNA structure prediction. A key focus is on the challenges these methods face before they become truly predictive, particularly in predicting emergent chemical phenomena. We believe that the ultimate goal of a simulation method or theory is to predict phenomena not seen before, and that Generative AI should be subject to these same standards before it is deemed useful for chemistry. We suggest that to overcome these challenges, future AI models need to integrate core chemical principles, especially from statistical mechanics.
Enhanced Sampling with Machine Learning: A Review
Jun 16, 2023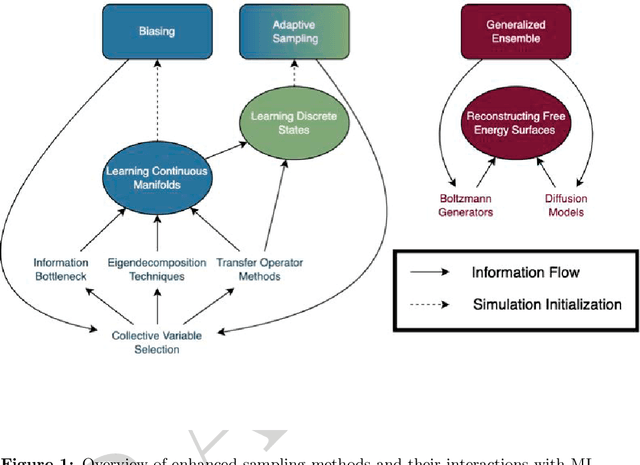



Molecular dynamics (MD) enables the study of physical systems with excellent spatiotemporal resolution but suffers from severe time-scale limitations. To address this, enhanced sampling methods have been developed to improve exploration of configurational space. However, implementing these is challenging and requires domain expertise. In recent years, integration of machine learning (ML) techniques in different domains has shown promise, prompting their adoption in enhanced sampling as well. Although ML is often employed in various fields primarily due to its data-driven nature, its integration with enhanced sampling is more natural with many common underlying synergies. This review explores the merging of ML and enhanced MD by presenting different shared viewpoints. It offers a comprehensive overview of this rapidly evolving field, which can be difficult to stay updated on. We highlight successful strategies like dimensionality reduction, reinforcement learning, and flow-based methods. Finally, we discuss open problems at the exciting ML-enhanced MD interface.