 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOrbital-free Bond Breaking via Machine Learning
Jun 07, 2013
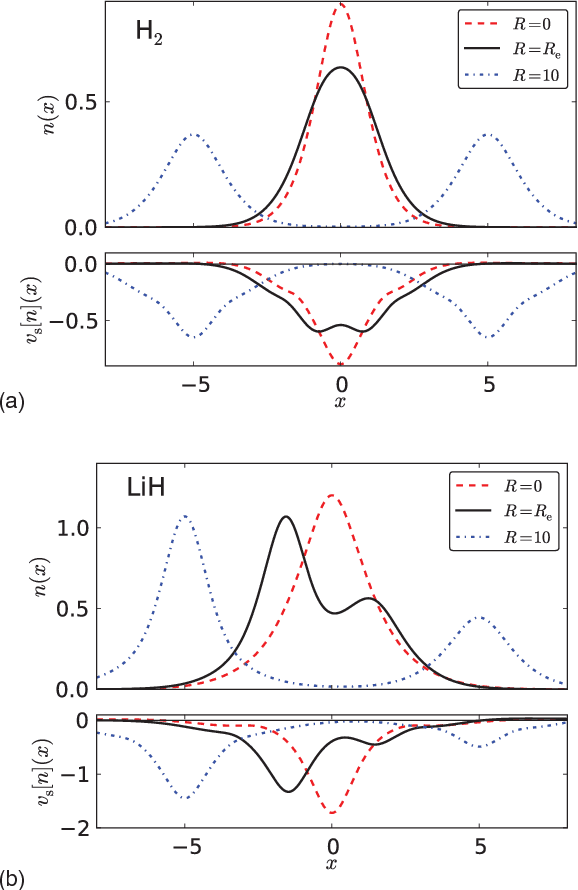

Machine learning is used to approximate the kinetic energy of one dimensional diatomics as a functional of the electron density. The functional can accurately dissociate a diatomic, and can be systematically improved with training. Highly accurate self-consistent densities and molecular forces are found, indicating the possibility for ab-initio molecular dynamics simulations.
Finding Density Functionals with Machine Learning
Dec 22, 2011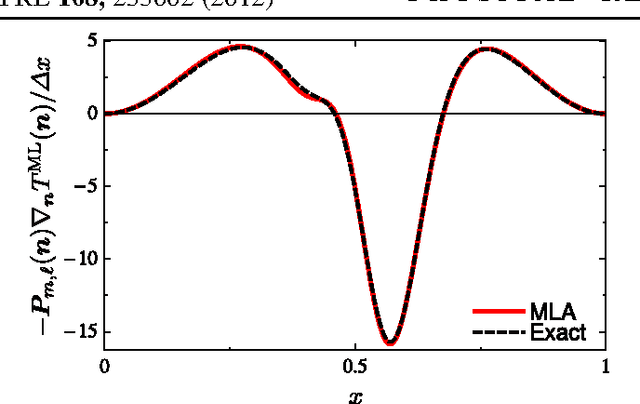
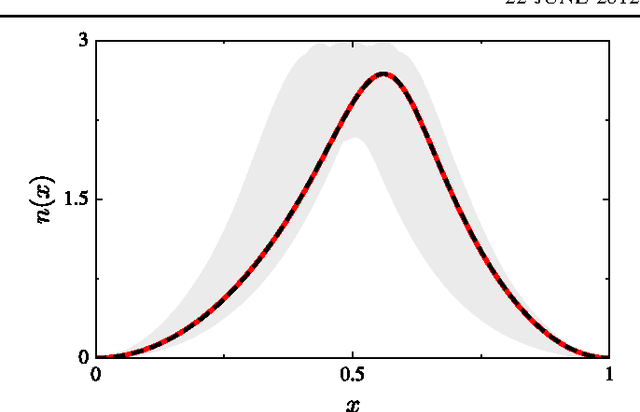
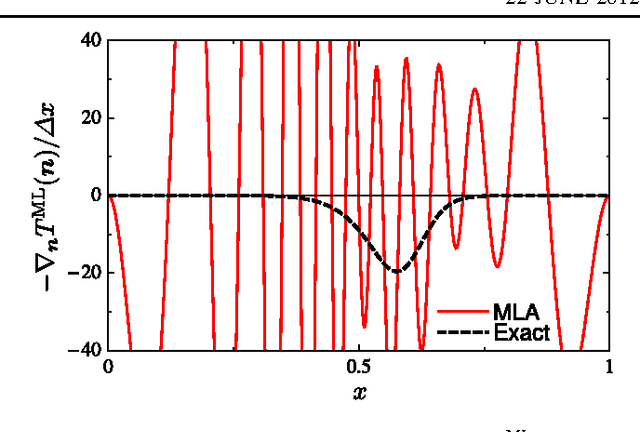
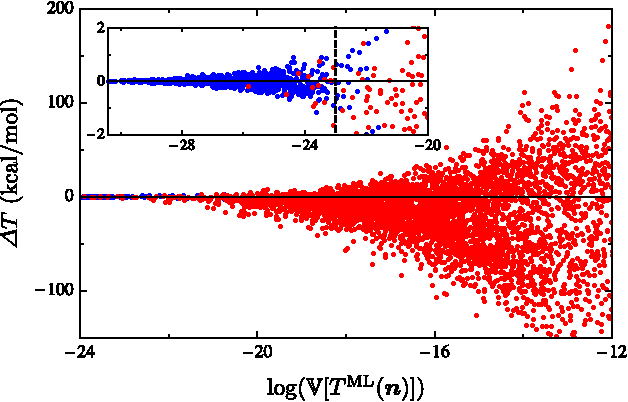
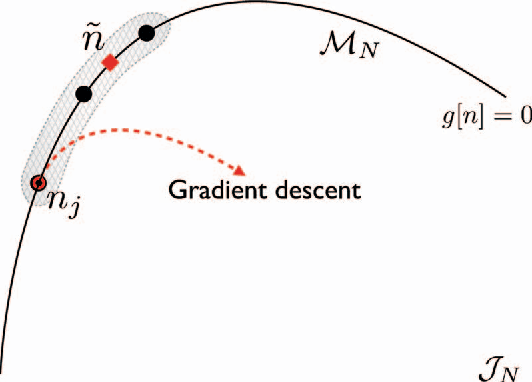
Machine learning is used to approximate density functionals. For the model problem of the kinetic energy of non-interacting fermions in 1d, mean absolute errors below 1 kcal/mol on test densities similar to the training set are reached with fewer than 100 training densities. A predictor identifies if a test density is within the interpolation region. Via principal component analysis, a projected functional derivative finds highly accurate self-consistent densities. Challenges for application of our method to real electronic structure problems are discussed.
How to Explain Individual Classification Decisions
Dec 06, 2009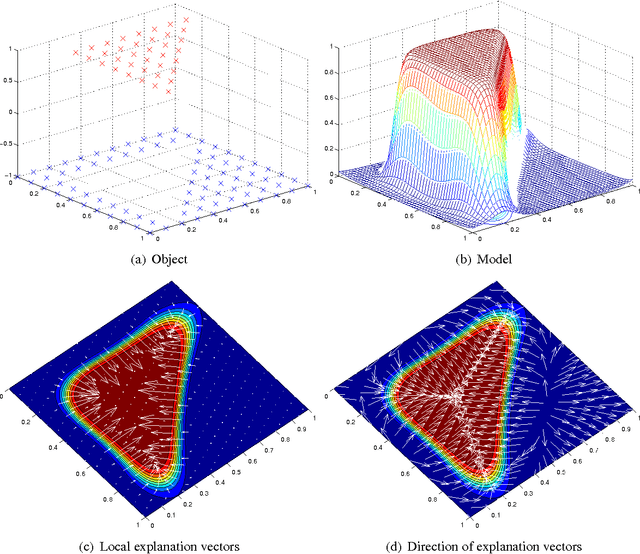
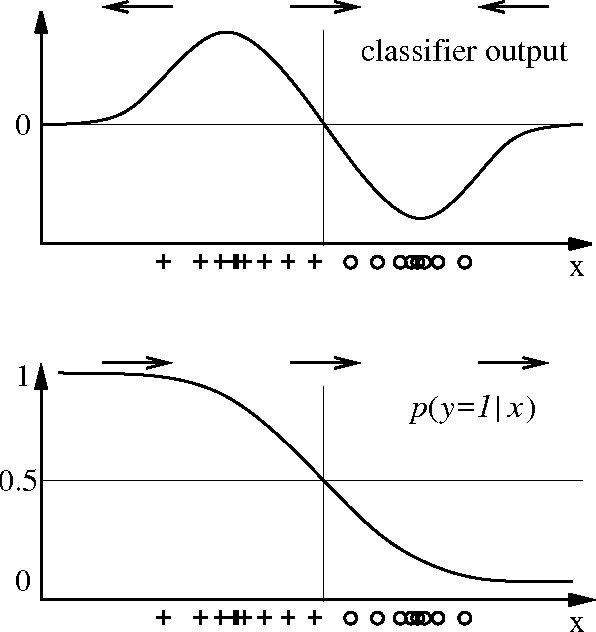

After building a classifier with modern tools of machine learning we typically have a black box at hand that is able to predict well for unseen data. Thus, we get an answer to the question what is the most likely label of a given unseen data point. However, most methods will provide no answer why the model predicted the particular label for a single instance and what features were most influential for that particular instance. The only method that is currently able to provide such explanations are decision trees. This paper proposes a procedure which (based on a set of assumptions) allows to explain the decisions of any classification method.