 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePretrained Joint Predictions for Scalable Batch Bayesian Optimization of Molecular Designs
Nov 14, 2025Batched synthesis and testing of molecular designs is the key bottleneck of drug development. There has been great interest in leveraging biomolecular foundation models as surrogates to accelerate this process. In this work, we show how to obtain scalable probabilistic surrogates of binding affinity for use in Batch Bayesian Optimization (Batch BO). This demands parallel acquisition functions that hedge between designs and the ability to rapidly sample from a joint predictive density to approximate them. Through the framework of Epistemic Neural Networks (ENNs), we obtain scalable joint predictive distributions of binding affinity on top of representations taken from large structure-informed models. Key to this work is an investigation into the importance of prior networks in ENNs and how to pretrain them on synthetic data to improve downstream performance in Batch BO. Their utility is demonstrated by rediscovering known potent EGFR inhibitors on a semi-synthetic benchmark in up to 5x fewer iterations, as well as potent inhibitors from a real-world small-molecule library in up to 10x fewer iterations, offering a promising solution for large-scale drug discovery applications.
Compressing physical properties of atomic species for improving predictive chemistry
Oct 31, 2018



The answers to many unsolved problems lie in the intractable chemical space of molecules and materials. Machine learning techniques are rapidly growing in popularity as a way to compress and explore chemical space efficiently. One of the most important aspects of machine learning techniques is representation through the feature vector, which should contain the most important descriptors necessary to make accurate predictions, not least of which is the atomic species in the molecule or material. In this work we introduce a compressed representation of physical properties for atomic species we call the elemental modes. The elemental modes provide an excellent representation by capturing many of the nuances of the periodic table and the similarity of atomic species. We apply the elemental modes to several different tasks for machine learning algorithms and show that they enable us to make improvements to these tasks even beyond simply achieving higher accuracy predictions.
Metadynamics for Training Neural Network Model Chemistries: a Competitive Assessment
Dec 19, 2017



Neural network (NN) model chemistries (MCs) promise to facilitate the accurate exploration of chemical space and simulation of large reactive systems. One important path to improving these models is to add layers of physical detail, especially long-range forces. At short range, however, these models are data driven and data limited. Little is systematically known about how data should be sampled, and `test data' chosen randomly from some sampling techniques can provide poor information about generality. If the sampling method is narrow `test error' can appear encouragingly tiny while the model fails catastrophically elsewhere. In this manuscript we competitively evaluate two common sampling methods: molecular dynamics (MD), normal-mode sampling (NMS) and one uncommon alternative, Metadynamics (MetaMD), for preparing training geometries. We show that MD is an inefficient sampling method in the sense that additional samples do not improve generality. We also show MetaMD is easily implemented in any NNMC software package with cost that scales linearly with the number of atoms in a sample molecule. MetaMD is a black-box way to ensure samples always reach out to new regions of chemical space, while remaining relevant to chemistry near $k_bT$. It is one cheap tool to address the issue of generalization.
The Many-Body Expansion Combined with Neural Networks
Sep 22, 2016
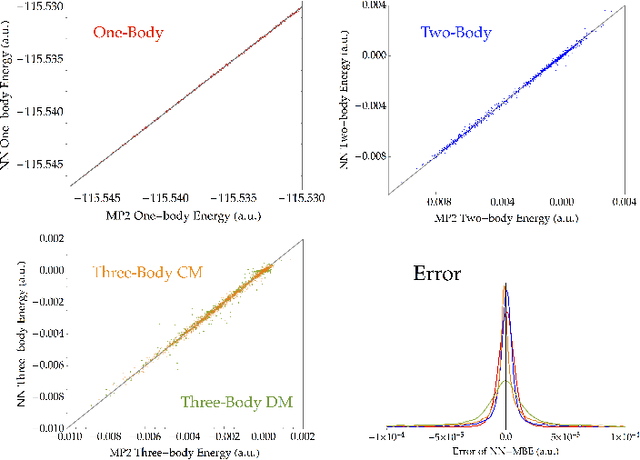
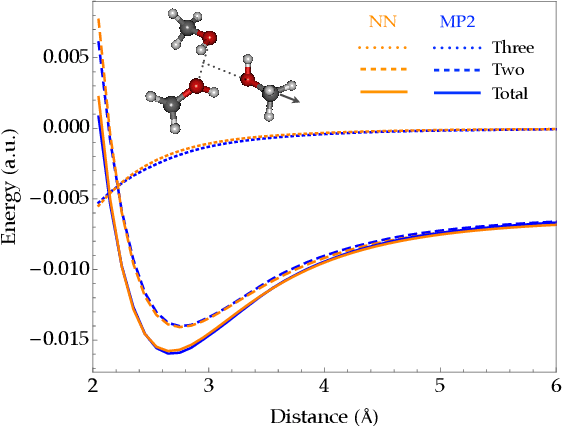
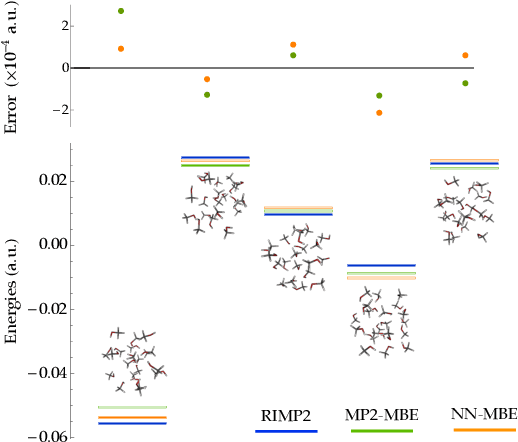
Fragmentation methods such as the many-body expansion (MBE) are a common strategy to model large systems by partitioning energies into a hierarchy of decreasingly significant contributions. The number of fragments required for chemical accuracy is still prohibitively expensive for ab-initio MBE to compete with force field approximations for applications beyond single-point energies. Alongside the MBE, empirical models of ab-initio potential energy surfaces have improved, especially non-linear models based on neural networks (NN) which can reproduce ab-initio potential energy surfaces rapidly and accurately. Although they are fast, NNs suffer from their own curse of dimensionality; they must be trained on a representative sample of chemical space. In this paper we examine the synergy of the MBE and NN's, and explore their complementarity. The MBE offers a systematic way to treat systems of arbitrary size and intelligently sample chemical space. NN's reduce, by a factor in excess of $10^6$ the computational overhead of the MBE and reproduce the accuracy of ab-initio calculations without specialized force fields. We show they are remarkably general, providing comparable accuracy with drastically different chemical embeddings. To assess this we test a new chemical embedding which can be inverted to predict molecules with desired properties.